Bone Marrow vs. Adipose Tissue: A Comparative Analysis of Mesenchymal Stem Cell Sources for Research and Therapy
This article provides a comprehensive, up-to-date analysis of mesenchymal stem cells (MSCs) derived from bone marrow and adipose tissue, the two most prevalent sources for research and clinical applications.
Bone Marrow vs. Adipose Tissue: A Comparative Analysis of Mesenchymal Stem Cell Sources for Research and Therapy
Abstract
This article provides a comprehensive, up-to-date analysis of mesenchymal stem cells (MSCs) derived from bone marrow and adipose tissue, the two most prevalent sources for research and clinical applications. Tailored for researchers, scientists, and drug development professionals, it systematically explores the fundamental biology, isolation methodologies, and characterization of BM-MSCs and AD-MSCs. It delves into their comparative differentiation potential, secretory profiles, and immunomodulatory properties, underpinned by direct comparative studies. The content further addresses critical challenges in the field, including donor variability, manufacturing standardization, and therapeutic efficacy, while highlighting advanced optimization strategies such as genetic engineering, preconditioning, and the emerging paradigm of cell-free therapies. This review synthesizes evidence to guide the selection and enhancement of MSC sources for specific therapeutic and regenerative applications.
The Biological Blueprint: Understanding MSC Origins and Fundamental Properties
The therapeutic potential of mesenchymal stromal cells (MSCs) has generated markedly increasing interest across a wide variety of biomedical disciplines, positioning them as cornerstone tools in regenerative medicine and immunomodulatory therapy [1] [2]. However, for years, investigators reported studies using different isolation methods, expansion protocols, and characterization approaches, creating significant challenges in comparing and contrasting research outcomes [1]. This heterogeneity threatened to hinder progress in the field, necessitating the development of standardized criteria.
To address this critical need, the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy (ISCT) established minimal criteria to define human MSCs, creating a foundational framework that has guided research and clinical development since its publication in 2006 [1]. This landmark position statement provided the scientific community with a unified reference point, enhancing the credibility and comparability of MSC studies worldwide [3]. The clarification of MSC nomenclature further refined scientific communication, establishing the distinction between "mesenchymal stromal cells" for the plastic-adherent population and reserving "mesenchymal stem cells" only for subsets meeting rigorous stem cell criteria [4].
This technical guide comprehensively details the ISCT criteria, explores their practical implementation in research settings, examines evolving standards in the field, and discusses the implications of recent advances in MSC biology for their definition and therapeutic application.
The ISCT Minimal Criteria: A Three-Pillar Framework
The ISCT established three fundamental criteria that must be satisfied to define human multipotent mesenchymal stromal cells [1]. These criteria serve as the cornerstone for MSC identification and characterization across diverse tissue sources and experimental applications.
Plastic Adherence Under Standard Culture Conditions
The first criterion requires that MSCs must be plastic-adherent when maintained in standard culture conditions [1] [3]. This fundamental property refers to the ability of MSCs to adhere to and proliferate on standard tissue culture plastic surfaces, forming characteristic fibroblast-like colonies. This adherence property enables the selective expansion of MSCs from heterogeneous cell mixtures obtained during tissue isolation procedures, serving as a primary purification step before further characterization.
Plastic adherence distinguishes MSCs from hematopoietic cells and other non-adherent cell populations present in source tissues like bone marrow, adipose tissue, or dental pulp. When placed in culture, MSCs attach to the plastic surface within 12-48 hours and begin to proliferate, eventually forming colonies that can be expanded through serial passaging while maintaining their fundamental characteristics.
Specific Surface Marker Expression Profile
The second criterion defines a specific immunophenotypic profile based on cluster of differentiation (CD) marker expression [1] [2]. According to ISCT standards, MSCs must demonstrate positive expression (≥95% of the population) of specific surface markers while simultaneously lacking expression (≤2% positive) of hematopoietic and monocytic markers.
Table 1: Required Surface Marker Profile for MSCs According to ISCT Criteria
| Marker Status | Surface Markers | Expression Requirement | Biological Significance |
|---|---|---|---|
| Positive Markers | CD105, CD73, CD90 | ≥95% positive | Identifies core MSC phenotype |
| Negative Markers | CD45, CD34, CD14 or CD11b, CD79α or CD19, HLA-DR | ≤2% positive | Excludes hematopoietic lineages |
CD105 (endoglin) is a type I membrane glycoprotein essential for cell migration and angiogenesis [2]. CD90 (Thy-1), an N-glycosylated glycosylphosphatidylinositol-anchored protein, mediates cell-cell and cell-extracellular matrix interactions, contributing to intercellular adhesion and migration [2]. CD73 functions as a 5'-exonuclease, catalyzing the hydrolysis of adenosine monophosphate into adenosine and playing a role in cell signaling within the bone marrow microenvironment [2].
The negative markers serve to exclude hematopoietic cells: CD45 is a marker for all white blood cells; CD34 identifies hematopoietic stem cells and endothelial cells; CD14/CD11b are expressed on monocytes and macrophages; CD79α/CD19 are markers of B cells; and HLA-DR is an MHC class II molecule found on antigen-presenting cells with strong immunogenic properties [2] [3].
Multilineage Differentiation Potential In Vitro
The third criterion requires that MSCs must demonstrate the capacity to differentiate into osteoblasts, adipocytes, and chondroblasts under standard in vitro inducing conditions [1]. This trilineage differentiation potential confirms the multipotent character of MSCs and provides functional validation of their stemness beyond surface marker expression.
Osteogenic differentiation is typically induced using culture medium supplemented with dexamethasone, ascorbic acid-2-phosphate, and β-glycerophosphate [5] [6]. Successful differentiation is confirmed by the formation of mineralized extracellular matrix detectable by Alizarin Red or Von Kossa staining.
Adipogenic differentiation requires induction with dexamethasone, isobutylmethylxanthine, indomethacin, and insulin [5] [6]. Differentiated adipocytes accumulate lipid vacuoles that can be visualized using Oil Red O staining.
Chondrogenic differentiation generally occurs in pellet or micromass culture systems with transforming growth factor-beta (TGF-β) supplementation [5] [6]. Successful chondrogenesis is demonstrated by the production of cartilage-specific extracellular matrix components detectable by Alcian Blue or Safranin O staining.
This trilineage differentiation capacity must be demonstrated under controlled in vitro conditions to meet the ISCT definition, providing functional evidence of multipotency that complements the immunophenotypic characterization.
Evolving Standards: Beyond the Minimal Criteria
While the ISCT minimal criteria established a crucial foundation, the field continues to evolve with emerging research revealing complexities in MSC biology that necessitate refined characterization approaches.
Tissue-Specific Variations and Functional Heterogeneity
Recent investigations have demonstrated that while MSCs from different sources share the core ISCT-defined characteristics, they exhibit significant biological variations based on their tissue of origin [5]. For instance, dental pulp-derived MSCs (DPSCs) consistently demonstrate smaller cell size, Nestin positivity, higher proliferation rates, and notably, a diminished capacity for adipogenic differentiation compared to adipose tissue-derived MSCs (ADSCs) [5]. This tissue-specific functional variation indicates that ontogeny significantly influences MSC properties, suggesting the need for tissue-specific characterization benchmarks alongside the core ISCT criteria.
Secretome analysis further reveals substantial differences in the profiles of anti-inflammatory and pro-inflammatory cytokines, chemokines, and growth factors between MSC populations from different tissues [5]. These variations extend to microRNA expression patterns, with DPSCs expressing microRNAs primarily involved in oxidative stress and apoptosis pathways, while ADSCs produce microRNAs that play regulatory roles in cell cycle and proliferation [5]. Such fundamental differences in secretory profiles have profound implications for selecting appropriate MSC sources for specific therapeutic applications.
Impact of Donor Physiology and Disease Status
The biological properties of MSCs are significantly influenced by donor characteristics, including age, health status, and disease conditions [6]. Comparative studies of ADSCs from healthy versus type 2 diabetic (T2D) donors reveal that while both populations meet the core ISCT criteria, they exhibit functional differences in specific differentiation capacities and pro-angiogenic potential [6]. Diabetic ADSCs demonstrate enhanced chondrogenic differentiation and pro-angiogenic properties compared to those from healthy donors, while showing reduced adipogenic differentiation potential [6].
These findings have crucial implications for autologous MSC therapies, particularly for patients with underlying metabolic conditions. They underscore the importance of donor-specific functional characterization beyond minimal criteria to predict therapeutic efficacy and inform clinical applications. The demonstration that diabetic MSCs retain significant functional capacity under diabetic culture conditions supports their potential use in autologous therapies for diabetic patients [6].
Advances in Characterization and Potency Assays
Contemporary MSC research increasingly recognizes the need for potency assays that reflect the intended mechanism of action in specific therapeutic contexts [7] [8]. While the ISCT minimal criteria focus on defining MSC identity, clinical translation requires demonstration of biological activity relevant to the target condition. Current analysis indicates that assessment of functionality remains limited in clinical trial reporting and does not always relate to the likely mechanism of action [7].
The ISCT continues to address these evolving needs through workshops and updated recommendations. A 2024 workshop on "Cell Therapies for Autoimmune Diseases: MSCs from Biology to Clinical Application" emphasized the need for standardization in design, conduct, and reporting of MSC clinical trials, including product characterization and key manufacturing parameters [8]. These developments highlight the ongoing evolution from minimal identity criteria toward comprehensive characterization frameworks that encompass identity, purity, viability, and clinically relevant potency measures.
MSC Characterization: Experimental Protocols and Methodologies
Standardized Workflow for MSC Characterization
The following diagram illustrates the comprehensive experimental workflow for isolating and characterizing MSCs according to ISCT criteria and contemporary standards:
Detailed Methodologies for Core Characterization Assays
Immunophenotyping by Flow Cytometry
Immunophenotypic analysis represents a critical component of MSC characterization, typically performed using flow cytometry. The standard protocol involves:
Cell Preparation: Harvest MSCs at 70-80% confluence (typically passages 3-6) using standard dissociation reagents like trypsin-EDTA [6]. Wash cells with phosphate-buffered saline (PBS) and adjust concentration to 1×10⁶ cells/mL in flow cytometry buffer.
Antibody Staining: Incubate cell aliquots with fluorochrome-conjugated antibodies against CD73, CD90, CD105, CD34, CD45, CD14, CD19, and HLA-DR for 30 minutes at 4°C in the dark [6]. Include appropriate isotype controls for compensation and background determination.
Analysis: Analyze stained cells using a flow cytometer, collecting a minimum of 10,000 events per sample. Evaluate positive marker expression (CD73, CD90, CD105) as ≥95% positive, while negative markers must demonstrate ≤2% expression to meet ISCT criteria [1] [3].
Trilineage Differentiation Assays
The functional multipotency of MSCs must be demonstrated through directed differentiation toward osteogenic, adipogenic, and chondrogenic lineages using established induction media [5] [6].
Table 2: Standardized Protocols for Trilineage Differentiation of MSCs
| Differentiation Pathway | Induction Media Components | Differentiation Period | Detection Method |
|---|---|---|---|
| Osteogenic | DMEM, 10% FBS, 50 µM ascorbic acid-2 phosphate, 10 mM β-glycerophosphate, 0.1 µM dexamethasone [5] | 21-28 days | Alizarin Red S staining for mineralized matrix |
| Adipogenic | DMEM, 10% FBS, 0.5 mM isobutylmethylxanthine, 1 µM dexamethasone, 10 µM insulin, 200 µM indomethacin [6] | 14-21 days | Oil Red O staining for lipid vacuoles |
| Chondrogenic | High-glucose DMEM, 1% ITS+ Premix, 50 µM ascorbic acid-2 phosphate, 0.1 µM dexamethasone, 10 ng/mL TGF-β3 [6] | 21-28 days | Alcian Blue staining for sulfated proteoglycans |
For osteogenic differentiation, seed MSCs at 3×10³ cells/well in 48-well plates and culture until 60-70% confluence before switching to osteogenic induction medium [5]. Replace differentiation medium every 3-4 days for 21-28 days. Fix cells with 4% paraformaldehyde and stain with 2% Alizarin Red S solution (pH 4.2) for 20 minutes to detect calcium deposits [6].
For adipogenic differentiation, culture confluent MSCs in adipogenic induction medium for 3 days followed by 1-3 days in maintenance medium, repeating this cycle for 2-3 weeks [6]. Fix cells and stain with filtered 0.3% Oil Red O solution in 60% isopropanol for 15 minutes to visualize lipid droplets.
For chondrogenic differentiation, pellet 2.5×10⁵ MSCs in conical polypropylene tubes and culture in chondrogenic induction medium with medium changes every 2-3 days [6]. After 21-28 days, fix pellets, embed in paraffin, section, and stain with 1% Alcian Blue in 3% acetic acid (pH 2.5) to detect sulfated glycosaminoglycans.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for MSC Characterization and Differentiation
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Culture Media | αMEM, DMEM, Basic Medium with supplements [5] [6] | MSC expansion and maintenance |
| Serum Supplements | Fetal Bovine Serum (FBS), Human Platelet Lysate (hPL) [5] [6] | Support MSC growth and proliferation |
| Dissociation Reagents | Trypsin-EDTA, TrypLE Select [5] [6] | Cell passaging and harvesting |
| Flow Cytometry Antibodies | Anti-CD73, CD90, CD105, CD34, CD45, CD14, CD19, HLA-DR [6] [3] | Immunophenotyping per ISCT criteria |
| Osteogenic Inducers | Ascorbic acid-2-phosphate, β-glycerophosphate, Dexamethasone [5] | Osteogenic differentiation |
| Adipogenic Inducers | Isobutylmethylxanthine, Dexamethasone, Insulin, Indomethacin [6] | Adipogenic differentiation |
| Chondrogenic Inducers | TGF-β3, ITS+ Premix, Dexamethasone [6] | Chondrogenic differentiation |
| Differentiation Stains | Alizarin Red S, Oil Red O, Alcian Blue [5] [6] | Detection of differentiated phenotypes |
Current Challenges and Future Perspectives
Gaps in Current Characterization Standards
Analysis of clinical trial reporting reveals significant limitations in MSC characterization, with approximately 33% of studies including no characterization data and only 13% reporting individual values per cell lot [7]. Viability was reported in just 57% of studies, while differentiation capacity was discussed for osteogenesis (29%), adipogenesis (27%), and chondrogenesis (20%) [7]. This substantial characterization deficit in clinical reporting highlights the disconnect between theoretical standards and practical implementation.
The extent of characterization in published trials shows no correlation with clinical phase of development, indicating that even late-phase trials often lack comprehensive product characterization [7]. Furthermore, assessment of functionality remains limited and frequently does not relate to the likely mechanism of action, representing a critical gap in the translational pathway [7].
Toward Next-Generation Characterization Standards
The emerging understanding of MSC heterogeneity and tissue-specific properties necessitates evolution beyond the minimal criteria toward more comprehensive characterization frameworks [5] [8]. Future standards will likely incorporate:
- Tissue-specific benchmarks that acknowledge biological variations while maintaining core identity criteria
- Mechanism-relevant potency assays that predict therapeutic efficacy for specific indications
- Secretome profiling to characterize paracrine activity, increasingly recognized as a primary therapeutic mechanism [5] [2]
- Standardized manufacturing protocols to control for process-induced variability [8]
- Donor screening criteria that account for health status and physiological factors influencing MSC function [6]
International standards organizations have begun addressing these needs through technical specifications for specific MSC types, such as the ISO/TS 24651:2022 for bone marrow-derived MSCs and ISO/TS 22859-1:2022 for umbilical cord-derived MSCs [3]. These documents outline requirements for collection, isolation, culture, characterization, quality control, and distribution, representing important steps toward global standardization.
The ISCT minimal criteria for defining MSCs have provided an essential foundation for standardizing research and clinical development in the field. The three pillars of plastic adherence, specific surface marker expression, and trilineage differentiation capacity continue to serve as the fundamental reference point for MSC identification across diverse tissue sources and applications. However, the evolving understanding of MSC biology reveals substantial tissue-specific variations, donor-dependent characteristics, and functional heterogeneity that necessitate refinement of characterization standards.
Future directions will require integration of core identity criteria with mechanism-relevant potency assays, comprehensive secretome profiling, and standardized manufacturing protocols to ensure predictive characterization and reliable therapeutic outcomes. As the field advances toward more personalized applications and precision medicine approaches, the definition of MSCs will continue to evolve, incorporating deeper understanding of their biological complexity while maintaining the standardized framework that enables scientific communication and progress.
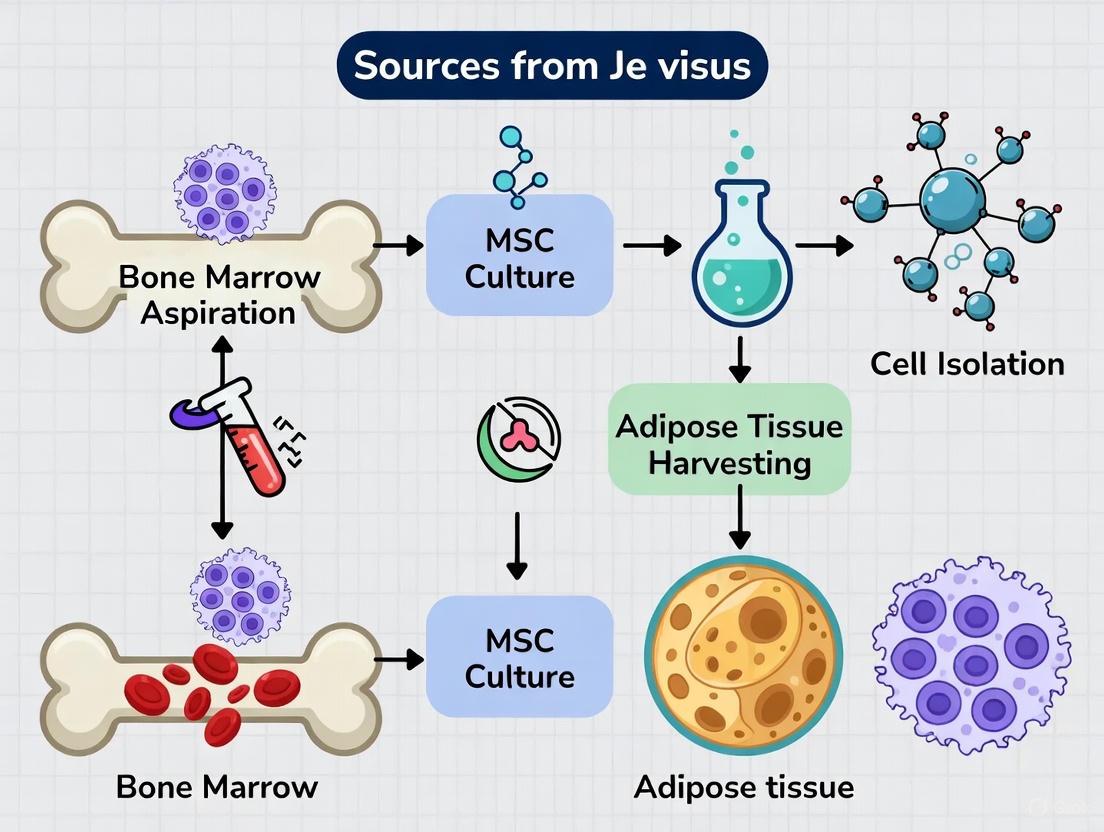
Mesenchymal Stromal Cells (MSCs) represent a cornerstone of regenerative medicine research due to their multipotent differentiation potential, immunomodulatory properties, and therapeutic versatility. While MSCs can be isolated from various tissues including adipose tissue, umbilical cord, and dental pulp, bone marrow-derived MSCs (BM-MSCs) remain the most extensively characterized and historically significant source [9]. First identified in the 1960s by Friedenstein and colleagues as plastic-adherent, fibroblast-like cells capable of forming bone colonies, BM-MSCs were the founding population that established the entire MSC field [10] [11]. The International Society for Cellular Therapy (ISCT) established minimal criteria for defining MSCs primarily based on BM-MSCs, cementing their role as the reference standard against which other MSC sources are compared [12] [9]. This technical guide provides an in-depth analysis of BM-MSC isolation, characterization, and fundamental biological properties within the broader context of MSC source comparison.
Fundamental Characteristics and Defining Criteria
BM-MSCs are multipotent stromal cells residing in the bone marrow niche, where they function as structural and regulatory components that support hematopoiesis [11]. According to ISCT standards, human BM-MSCs must fulfill three minimal criteria:
- Plastic adherence under standard culture conditions
- Specific surface marker expression: Positive for CD105, CD73, CD90; negative for CD45, CD34, CD14/CD11b, CD79α/CD19, and HLA-DR
- Multilineage differentiation potential: Ability to differentiate into osteoblasts, adipocytes, and chondroblasts in vitro [12] [9]
Beyond these minimal criteria, BM-MSCs exhibit distinctive biological properties that position them uniquely among MSC sources. They demonstrate a characteristic spindle-shaped, fibroblastic morphology with extensions projecting from a small cell body [12] [10]. While adipose tissue contains a higher frequency of MSCs per unit volume, BM-MSCs are renowned for their robust osteogenic differentiation capacity, which often exceeds that of adipose-derived MSCs (AD-MSCs) [11]. BM-MSCs also possess potent immunomodulatory functions, influencing both innate and adaptive immune responses through cell-cell contact and secretion of bioactive factors [12] [13].
Table 1: Key Surface Markers for Identifying Human BM-MSCs
| Marker Category | Marker Examples | Expression in BM-MSCs | Functional Significance |
|---|---|---|---|
| Positive Markers | CD73, CD90, CD105 | Positive | Mesenchymal lineage commitment; ectoenzyme activity |
| CD29, CD44, CD166 | Positive | Cell adhesion and migration | |
| CD146, STRO-1 | Positive (subsets) | Primitive/perivascular phenotype | |
| Negative Markers | CD34, CD45 | Negative | Exclusion of hematopoietic lineage |
| CD14, CD11b | Negative | Exclusion of monocyte/macrophage lineage | |
| CD19, CD79α | Negative | Exclusion of B-cell lineage | |
| HLA-DR | Negative (unless stimulated) | Exclusion of activated immune cells |
Isolation Protocols and Methodologies
Bone Marrow Aspiration and Initial Processing
Bone marrow for BM-MSC isolation is typically obtained from the iliac crest, vertebral body, sternum, or femoral shaft [10]. For human isolation, iliac crest aspiration is most common, performed aseptically after obtaining informed consent [12]. The basic workflow begins with collection of bone marrow into anticoagulant-treated tubes, followed by centrifugation to separate the buffy coat from red blood cells and plasma [12].
Core Isolation Techniques
Several methods have been established for isolating BM-MSCs from the initial marrow aspirate:
Density Gradient Centrifugation This represents the most widely used isolation technique, providing effective separation of mononuclear cells from erythrocytes and granulocytes [12] [9].
- Protocol: The buffy coat or diluted bone marrow is carefully layered onto an equal volume of density gradient medium (Ficoll-Paque or Percoll)
- Centrifugation: 400-450 × g for 20-30 minutes at room temperature
- Cell recovery: Mononuclear cells forming a distinct interface ring are collected, washed twice in phosphate-buffered saline (PBS), and counted
- Advantages: Effective removal of erythrocytes and granulocytes; high cell viability
- Disadvantages: Can co-isolate other mononuclear cells requiring further purification [12]
Plastic Adherence Selection This method exploits the fundamental property of MSC adhesion to tissue culture plastic, providing a simple purification approach that can be used alone or following density gradient separation [9] [11].
- Protocol: Washed mononuclear cells are plated on standard tissue culture-treated flasks or plates
- Culture medium: Typically Dulbecco's Modified Eagle Medium (DMEM) or αMEM supplemented with 10% fetal bovine serum (FBS) or human platelet lysate and antibiotics
- Incubation conditions: 37°C in a humidified atmosphere with 5% CO₂ for 48 hours
- Medium exchange: Non-adherent cells (primarily hematopoietic) are removed after 48-72 hours by PBS washing; medium is exchanged every 3-4 days thereafter
- Advantages: Technically simple; no specialized equipment required
- Disadvantages: Slow selection process; potential hematopoietic cell contamination in early passages [12] [11]
Enzymatic Digestion Approach For bone marrow fragments or whole bone samples, enzymatic digestion liberates stromal cells from the matrix [10].
- Protocol: Marrow plugs or bone fragments are treated with collagenase type I or II (typically 1-3 mg/mL) for 30-60 minutes at 37°C
- Termination: Enzyme activity is neutralized with serum-containing medium
- Cell recovery: Released cells are collected by centrifugation and plated
- Advantages: Higher yield from compact bone sources; preserves perivascular MSC populations
- Disadvantages: Potential enzyme-induced cell surface marker alteration; requires optimization [10]
Immunodepletion or Positive Selection Advanced isolation employs antibody-based strategies to deplete hematopoietic cells (using anti-CD45, anti-CD34) or positively select MSC populations (using anti-STRO-1, anti-CD271, anti-CD106) via fluorescence-activated cell sorting (FACS) or magnetic-activated cell sorting (MACS) [11].
- Advantages: Highly pure populations; ability to select specific MSC subpopulations
- Disadvantages: Technically demanding; expensive; requires specialized equipment [11]
Diagram 1: BM-MSC isolation workflow. The process begins with bone marrow aspiration, followed by various isolation methods that converge on culture expansion and final characterization.
Characterization and Functional Validation
Morphological Assessment
BM-MSCs exhibit a characteristic homogeneous fibroblastic morphology with a small cell body and extensions projecting in opposite directions [12] [10]. Under phase-contrast microscopy, early passage cells appear heterogeneous with distinct colony formation, gradually developing a more uniform spindle-shaped appearance with increased proliferation [12]. Murine BM-MSCs are notably smaller than human, canine, and feline BM-MSCs, demonstrating species-specific morphological variations [10].
Immunophenotyping by Flow Cytometry
Comprehensive surface marker analysis is essential for BM-MSC identification and quality control. Standard protocol involves:
- Cell preparation: Harvest P2-P3 cells at 80-90% confluence using enzymatic detachment; wash with PBS containing 1% BSA
- Antibody staining: Incubate 10⁵ cells/tube with fluorochrome-conjugated antibodies against CD73, CD90, CD105, CD34, CD45, CD14, and appropriate isotype controls
- Analysis: Acquire data on flow cytometer; analyze ≥10,000 events using appropriate gating strategies
- Interpretation: ≥95% positive for CD73, CD90, CD105; ≤2% positive for hematopoietic markers [12]
Table 2: Growth Kinetics and Senescence of BM-MSCs from Different Species
| Species | Population Doubling Time (PDT) | Senescence Characteristics | Proliferation Marker Expression |
|---|---|---|---|
| Human | <48 hours (early passage, P1-P5) >96 hours (late passage, P6+) | SA-β-Gal presence in late passages (P6+) | Ki67 negative in cultured cells |
| Canine | Increases to ~100 hours after 25 days | SA-β-Gal increases with passage number | Ki67 positive |
| Rat | 20-30 hours (P1-P3) 50-130 hours (P4-P5) | SA-β-Gal absent at PD100 Present in P6 | Ki67 positive |
| Mouse | >80 hours at week 4 and 8 (P2) | Low senescence in P3-P4 | Information limited |
Trilineage Differentiation Potential
Functional validation of BM-MSCs requires demonstration of adipogenic, osteogenic, and chondrogenic differentiation capacity using specific induction media.
Adipogenic Differentiation Protocol
- Induction: Culture confluent BM-MSCs for 1-3 weeks in adipogenic differentiation medium (typically containing insulin, dexamethasone, indomethacin, and IBMX)
- Staining: Fix cells with 4% formaldehyde; stain lipid droplets with Oil Red O (0.5% in isopropanol) for 10-15 minutes
- Visualization: Microscopic observation of red-stained intracellular lipid vacuoles
- Quantification: Oil Red O elution with 100% isopropanol and spectrophotometric measurement at 510 nm [12]
Osteogenic Differentiation Protocol
- Induction: Culture confluent BM-MSCs for 3-4 weeks in osteogenic medium (typically containing dexamethasone, ascorbate-2-phosphate, and β-glycerophosphate)
- Staining: Fix with 4% formaldehyde; stain calcium deposits with 2% Alizarin Red S (pH 4.1-4.3) for 5-10 minutes
- Visualization: Microscopic observation of orange-red mineralized nodules
- Quantification: Alizarin Red extraction with 10% cetylpyridinium chloride and spectrophotometric measurement at 562 nm [12] [14]
Chondrogenic Differentiation Protocol
- Induction: Pellet or micromass culture of 2.5×10⁵ BM-MSCs in chondrogenic medium (typically containing TGF-β3, dexamethasone, ascorbate-2-phosphate, proline, and insulin-transferrin-selenium)
- Duration: 3-4 weeks with medium changes every 2-3 days
- Staining: Fix pellets with 4% formaldehyde; embed in paraffin; section; stain with 1% Alcian Blue (pH 2.5) for 30 minutes
- Visualization: Microscopic observation of blue-stained proteoglycan-rich extracellular matrix [15]
Diagram 2: BM-MSC characterization workflow. The validation process requires confirmation of morphological features, specific surface marker expression, and demonstrated trilineage differentiation potential.
Culture Conditions and Expansion Considerations
Media Formulations and Supplements
Traditional BM-MSC expansion employs basal media (DMEM or αMEM) supplemented with 10% FBS, which provides essential growth factors and attachment factors but introduces batch-to-batch variability and xenogenic risks [13]. For clinical applications, serum-free media (SFM) and xeno-free media (XFM) have been developed using defined components including recombinant growth factors, lipids, and proteins [13]. Human platelet lysate (hPL) has emerged as an effective FBS alternative, promoting superior proliferation while eliminating xenogenic concerns [13].
Seeding Density and Population Doubling
BM-MSCs exhibit density-dependent growth inhibition and should be seeded at 1,000-5,000 cells/cm² for optimal expansion [13]. Population doubling time (PDT) varies by species and passage number, with human BM-MSCs typically demonstrating PDT of <48 hours in early passages (P1-P5) increasing to >96 hours in later passages (P6+) [10]. Serial passaging beyond P6-8 often leads to replicative senescence characterized by enlarged morphology, slowed proliferation, and increased SA-β-Gal activity [10].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for BM-MSC Isolation and Characterization
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Isolation Reagents | Ficoll-Paque Percoll Collagenase Type I/II Antibodies (CD45, CD34) | Density gradient separation Density gradient separation Enzymatic digestion of bone fragments Hematopoietic cell depletion |
| Culture Media & Supplements | αMEM/DMEM Fetal Bovine Serum (FBS) Human Platelet Lysate (hPL) Penicillin/Streptomycin | Basal culture medium Traditional growth supplement Xeno-free growth supplement Antibiotic prevention of contamination |
| Characterization Reagents | Anti-CD73, CD90, CD105 Anti-CD34, CD45, CD14 Oil Red O Alizarin Red S Alcian Blue | Positive marker flow cytometry Negative marker flow cytometry Adipogenic differentiation staining Osteogenic differentiation staining Chondrogenic differentiation staining |
| Differentiation Kits | StemPro Adipogenesis Kit StemPro Osteogenesis Kit StemPro Chondrogenesis Kit | Standardized adipogenic differentiation Standardized osteogenic differentiation Standardized chondrogenic differentiation |
When positioned within the broader landscape of MSC sources, BM-MSCs demonstrate distinct advantages and limitations. Adipose tissue provides approximately 500 times more MSCs per unit volume than bone marrow, and extraction via liposuction is considered less invasive than bone marrow aspiration [15]. However, BM-MSCs possess superior documented clinical history with more extensive clinical trial data across various disease indications [16]. The biological performance also varies, with AD-MSCs demonstrating enhanced angiogenic potential in some diabetic models, while BM-MSCs maintain robust osteogenic capability [15] [11]. Importantly, bone marrow aspiration collects additional beneficial cell populations including hematopoietic stem cells and endothelial progenitor cells, which are absent in adipose-derived isolates [16].
BM-MSCs represent the foundational reference standard for mesenchymal stromal cell research, characterized by well-defined isolation protocols, comprehensive characterization criteria, and extensive clinical validation. Their robust osteogenic potential, documented safety profile, and historical precedence continue to make them invaluable for regenerative medicine applications. While newer MSC sources offer advantages in terms of accessibility and abundance, BM-MSCs remain essential for both basic research and clinical development, providing the benchmark against which alternative MSC sources are evaluated. As the field advances toward serum-free culture conditions and more precise subpopulation identification, BM-MSCs will continue to be a critical tool for understanding MSC biology and therapeutic mechanisms.
Adipose-derived mesenchymal stem cells (AD-MSCs), also referred to as adipose-derived stromal cells or adipose-derived stem cells (ASCs), are a population of multipotent progenitor cells residing in adipose tissue. They are a subtype of mesenchymal stem cells (MSCs) that have garnered significant interest in regenerative medicine due to their accessibility, abundance, and robust regenerative capabilities [17] [18]. According to the latest recommendations from the International Society for Cell & Gene Therapy (ISCT), these cells should be precisely identified by their tissue of origin [19]. AD-MSCs are defined by a triad of key characteristics: their ability to adhere to plastic surfaces under standard culture conditions, specific surface marker expression, and trilineage differentiation potential into adipocytes, osteoblasts, and chondrocytes in vitro [2] [20].
Within the broader thesis on MSC sources, AD-MSCs present a compelling alternative to the more traditionally studied bone marrow-derived MSCs (BM-MSCs). While BM-MSCs were the first discovered and are the most extensively characterized, AD-MSCs offer distinct practical advantages, primarily their ease of harvest from abundant adipose tissue and a less invasive isolation procedure, often yielding a higher number of cells per gram of starting tissue [18] [20]. This positions AD-MSCs as a highly viable and efficient cell source for research and clinical applications in tissue engineering and regenerative medicine [21].
Isolation and Culture Protocols
The isolation of AD-MSCs primarily relies on the enzymatic digestion of adipose tissue, a method that allows for the release of the stromal vascular fraction (SVF) containing the stem cells.
Core Isolation Methodology
The standardized protocol for isolating AD-MSCs from human adipose tissue involves a series of critical steps to obtain a viable and pure cell population [19] [22].
Table: Key Steps in the Isolation of Human AD-MSCs
| Step | Description | Key Reagents/Equipment |
|---|---|---|
| 1. Tissue Harvesting | Adipose tissue is obtained via liposuction (e.g., power-assisted) or surgical resection from subcutaneous deposits [19]. | -- |
| 2. Washing & Mincing | Tissue is washed with physiological saline or PBS to remove blood and contaminants, then minced into small fragments [22]. | Phosphate-Buffered Saline (PBS), Antibiotics (e.g., Penicillin/Streptomycin) |
| 3. Enzymatic Digestion | Minced tissue is digested with collagenase (e.g., Type I or II) at 37°C for 30-60 minutes with agitation [19] [22]. | Collagenase Type I/II, Dulbecco's Modified Eagle Medium (DMEM), Serum |
| 4. Digestion Neutralization | Enzyme activity is halted by adding culture medium supplemented with serum [19] [22]. | Fetal Bovine Serum (FBS) |
| 5. Centrifugation | The digest is centrifuged to separate the stromal vascular fraction (SVF) pellet from mature adipocytes and lipids [19] [22]. | Centrifuge |
| 6. Erythrocyte Lysis & Filtration | The SVF pellet is treated with NH4Cl to lyse red blood cells and filtered through a 100 μm sieve [19]. | NH4Cl solution, Cell strainer (70-100 μm) |
| 7. Plating & Culture | The processed SVF is resuspended in growth medium and plated on a tissue culture flask [22]. | Culture flask, DMEM, FBS, Antibiotics |
Experimental Workflow Visualization
The following diagram illustrates the complete isolation and initial characterization workflow for AD-MSCs:
Characterization and Key Properties
Rigorous characterization is essential to confirm the identity and quality of isolated AD-MSCs, following international standards.
Surface Marker Profile
AD-MSCs are defined by a specific immunophenotype. The following table summarizes the positive and negative markers used for their identification via flow cytometry [19] [2] [20].
Table: Standard Surface Marker Profile for Human AD-MSCs
| Marker Category | Specific Markers | Presence | Significance / Function |
|---|---|---|---|
| Positive Markers | CD73, CD90, CD105 | ≥ 95% Expression | Core defining markers per ISCT; involved in purinergic signaling (CD73), cell-cell/matrix adhesion (CD90), and angiogenesis (CD105) [2]. |
| Negative Markers | CD11b, CD14, CD19, CD34, CD45, HLA-DR | ≤ 2% Expression | Absence of hematopoietic (CD11b, CD14, CD19, CD34, CD45) and potent immunogenic (HLA-DR) markers [19] [2]. |
| Other Common Markers | CD29, CD44, CD49d | Variable Expression | Associated with adhesion and migration [20]. |
| Other Negative Markers | Stro-1 | Low/Absent Expression | A marker often found on BM-MSCs, typically low on AD-MSCs [20]. |
Trilineage Differentiation Potential
A functional hallmark of AD-MSCs is their ability to differentiate into multiple mesodermal lineages. The standard protocol involves culturing the cells in specific inductive media [22] [20].
Table: Standard Protocols for Trilineage Differentiation of AD-MSCs
| Lineage | Key Inductive Factors | Differentiation Timeline | Common Verification Methods |
|---|---|---|---|
| Adipogenic | Dexamethasone, Indomethacin, IBMX, Insulin [20]. | 14-21 days | Oil Red O staining of lipid vacuoles [22] [20]. |
| Osteogenic | Dexamethasone, Ascorbate-2-phosphate, β-Glycerophosphate [20]. | 14-21 days | Alizarin Red S staining of calcium deposits, Alkaline Phosphatase (ALP) activity assay [20]. |
| Chondrogenic | TGF-β (e.g., TGF-β3), Dexamethasone, Ascorbate-2-phosphate, Proline [20]. | 21-28 days | Alcian Blue or Toluidine Blue staining of sulfated proteoglycans in pellet or micromass culture [20]. |
Functional Properties and Comparison with Other MSCs
AD-MSCs are not merely defined by their surface markers but by their potent functional capacities, which are central to their therapeutic application.
Paracrine Signaling and Exosome Biology
The therapeutic effects of AD-MSCs are now largely attributed to their paracrine activity rather than direct cell replacement [19] [18]. They secrete a complex mixture of bioactive molecules, collectively known as the secretome, which includes growth factors, cytokines, chemokines, and extracellular vesicles (EVs) like exosomes [19]. AD-MSC-derived exosomes carry proteins, lipids, and nucleic acids (e.g., miRNAs) that can modulate recipient cell behavior, promoting processes such as angiogenesis, immunomodulation, and tissue repair [18]. This has led to the emergence of "stem cell-free" therapies utilizing the AD-MSC secretome or exosomes as a potentially safer and more controllable alternative to whole-cell transplants [19] [23].
Key Signaling Pathways
The regenerative and immunomodulatory functions of AD-MSCs are mediated through the activation or inhibition of multiple intracellular signaling pathways [21] [18].
AD-MSCs vs. Bone Marrow MSCs (BM-MSCs)
A donor-matched comparison of AD-MSCs and BM-MSCs reveals both similarities and critical, tissue-specific differences that influence their application [20].
Table: Donor-Matched Comparison of AD-MSCs and BM-MSCs
| Property | AD-MSCs | BM-MSCs | Research Implications |
|---|---|---|---|
| Tissue Source & Accessibility | Abundant subcutaneous fat; minimally invasive harvest (liposuction) [18] [20]. | Iliac crest aspiration; more invasive and painful harvest [20]. | AD-MSCs are often preferred for autologous therapy due to easier access. |
| Cell Yield | High yield of cells per gram of tissue [18]. | Low frequency (0.001-0.01%) in bone marrow aspirate [20]. | AD-MSCs may require less in vitro expansion. |
| Proliferation Rate | Significantly higher proliferation capacity [20]. | Lower proliferation rate [20]. | AD-MSCs can be expanded more rapidly for clinical use. |
| Osteogenic Potential | Moderate | Superior (earlier and higher ALP activity, calcium deposition) [20]. | BM-MSCs may be preferred for bone tissue engineering. |
| Chondrogenic Potential | Moderate | Superior [20]. | BM-MSCs may be preferred for cartilage repair. |
| Adipogenic Potential | Superior (high lipid vesicle formation) [20]. | Moderate | AD-MSCs are inherently primed for adipogenesis. |
| Specific Marker Expression | High CD49d, Low Stro-1 [20]. | Low CD49d, High Stro-1 [20]. | Useful for distinguishing cell origin. |
The Scientist's Toolkit: Essential Research Reagents
Successful isolation, culture, and differentiation of AD-MSCs require a suite of validated reagents and materials.
Table: Essential Research Reagents for AD-MSC Work
| Reagent / Material | Function / Purpose | Specific Examples |
|---|---|---|
| Collagenase Type I/II | Enzymatic digestion of the extracellular matrix in adipose tissue to release the Stromal Vascular Fraction (SVF) [22]. | Collagenase from Clostridium histolyticum |
| Dulbecco's Modified Eagle Medium (DMEM) | Base nutrient medium for cell culture and expansion [22]. | Low-glucose DMEM |
| Fetal Bovine Serum (FBS) | Essential supplement for cell culture media, providing growth factors, hormones, and attachment factors [22]. | Qualified, lot-tested FBS |
| Flow Cytometry Antibodies | Characterization of cell surface markers to confirm AD-MSC identity (positive and negative markers) [20]. | Antibodies against CD73, CD90, CD105, CD34, CD45, HLA-DR |
| Trilineage Differentiation Kits | Defined media supplements to induce and support differentiation into adipocytes, osteocytes, and chondrocytes [20]. | StemPro Differentiation Kits |
| Trypsin/EDTA | Enzymatic detachment of adherent cells from culture plastic for passaging and sub-culturing [20]. | 0.25% Trypsin-EDTA |
| Dexamethasone | A synthetic glucocorticoid used as a key inductive factor in adipogenic, osteogenic, and chondrogenic differentiation media [20]. | -- |
The field of regenerative medicine has long relied on bone marrow (BM) and adipose tissue (AT) as primary sources for mesenchymal stem cells (MSCs). These cells are defined by their adherence to plastic, specific surface marker expression (CD73+, CD90+, CD105+, CD34-, CD45-, HLA-DR-), and trilineage differentiation potential [2] [3]. However, the search for more accessible, potent, and less immunogenic alternatives has led researchers to investigate perinatal and adult biological materials previously considered medical waste. Umbilical cord (UC), placenta (PL), and menstrual blood-derived MSCs (MenSCs) represent promising alternatives with distinct biological advantages [3] [24] [25]. This review provides a technical comparison of these emerging MSC sources, detailing their isolation, characterization, and therapeutic mechanisms for researchers and drug development professionals.
Source-Specific Characterization and Isolation Protocols
Umbilical Cord Mesenchymal Stem Cells (UC-MSCs)
UC-MSCs are typically isolated from Wharton's jelly, the mucoid connective tissue surrounding the umbilical cord vessels [3]. This source provides a high concentration of MSCs with enhanced proliferative capacity compared to their bone marrow-derived counterparts [26].
Isolation Protocol:
- Sample Preparation: Obtain informed consent and collect umbilical cord tissue (approx. 10-15 cm) post-delivery under sterile conditions. Transport in cold (4°C) saline solution supplemented with antibiotics (e.g., Penicillin-Streptomycin).
- Processing: Dissect cord segments to remove blood vessels. Mechanically mince Wharton's jelly into 2-3 mm³ explants.
- Explant Culture: Plate explants in culture-treated flasks with Dulbecco's Modified Eagle Medium (DMEM)/F12 supplemented with 10-15% Fetal Bovine Serum (FBS), 1% L-glutamine, and 1% antibiotic-antimycotic solution.
- Outgrowth and Expansion: Maintain at 37°C with 5% CO₂. MSC outgrowth typically occurs within 7-14 days. Passage cells at 80-90% confluence using 0.25% trypsin-EDTA [3] [26].
Placental Mesenchymal Stem Cells (PMSCs)
The placenta contains MSCs within its amnion, chorionic frondosum, and basal decidua regions [3]. PMSCs may exhibit superior proliferative capacity compared to UC-MSCs and demonstrate pronounced immunosuppressive effects on dendritic cells and T cells [3].
Isolation Protocol:
- Tissue Processing: Collect placental tissue under sterile conditions. Separate amnion and chorion layers by blunt dissection.
- Enzymatic Digestion: Mince tissues and digest with 2 mg/mL collagenase type IV and 0.5 mg/mL hyaluronidase for 3-4 hours at 37°C with agitation.
- Cell Recovery: Neutralize enzymes with complete culture medium. Filter cell suspension through 100μm and 70μm cell strainers.
- Culture Establishment: Plate cells at 5×10⁴ cells/cm² in α-MEM supplemented with 10% FBS, 5 ng/mL bFGF, and antibiotics. Passage at 80% confluence [3].
Menstrual Blood-Derived Mesenchymal Stem Cells (MenSCs)
MenSCs are collected from menstrual effluent containing cellular material shed from the functionalis layer of the endometrium [27] [25]. These cells demonstrate a remarkably high proliferation rate, doubling approximately every 19.4 hours compared to 40-45 hours for BM-MSCs [27].
Isolation Protocol:
- Sample Collection: Utilize menstrual cup (e.g., DivaCup) or specialized collection device (e.g., Instead SoftCup) during first 2-3 days of menstruation.
- Transport Medium: Collect in sterile containers with transport medium containing antibiotics and antifungals.
- Processing: Dilute menstrual blood 1:1 with phosphate-buffered saline (PBS). Separate mononuclear cells using Ficoll-Paque density gradient centrifugation (800×g for 30 minutes).
- Culture Establishment: Plate cells at 1×10⁵ cells/cm² in DMEM/F12 with 10% FBS, 1% antibiotic-antimycotic. Maintain at 37°C with 5% CO₂. First medium change after 48 hours to remove non-adherent cells [27] [25] [28].
Table 1: Comparative Analysis of Alternative MSC Sources
| Characteristic | UC-MSCs | PMSCs | MenSCs |
|---|---|---|---|
| Primary Tissue Source | Wharton's Jelly | Amnion, Chorion, Decidua | Endometrial Functional Layer |
| Cell Yield | High (0.5-2×10⁶ cells/cm cord) | Moderate-High (Varies by region) | Moderate (1-5×10⁵ cells/mL effluent) |
| Population Doubling Time | ~30 hours | ~30-40 hours | ~19.4 hours |
| Unique Surface Markers | CD146+ [26] | HLA-G+ (reported in some studies) | Oct-4+, SOX2+, NANOG+ [27] |
| Key Secretory Factors | High HGF, FGF-2, NGF [26] | TGF-β, PGE2 | MMPs, VEGF, Angiogenin |
| Cryopreservation Recovery | >85% | >80% | >90% |
| Special Considerations | ISO/TS 22859-1:2022 standards [3] | Complex tissue composition requires careful isolation | Cyclical availability, can be collected repeatedly |
Experimental Workflow for MSC Characterization
The following diagram illustrates the standard experimental workflow for isolating and characterizing MSCs from alternative sources:
Diagram 1: MSC Isolation and Characterization Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Alternative MSC Studies
| Reagent/Category | Specific Examples | Research Function | Application Notes |
|---|---|---|---|
| Culture Media | DMEM/F12, α-MEM, MSC-Qualified FBS | Supports MSC expansion while maintaining differentiation potential | UC-MSCs often require specialized supplements (bFGF) for optimal growth [26] |
| Dissociation Reagents | Collagenase Type IV, Hyaluronidase, Trypsin-EDTA | Tissue dissociation and cell passaging | Enzyme concentration and duration vary by tissue source (higher for placenta) |
| Characterization Antibodies | CD73, CD90, CD105, CD34, CD45, HLA-DR | Flow cytometry immunophenotyping | Essential for ISCT compliance; MenSCs require additional pluripotency markers [2] [27] |
| Differentiation Kits | Osteo-, Chondro-, Adipogenic Induction Media | Trilineage differentiation potential assessment | Standardized kits ensure reproducible differentiation across laboratories |
| Extracellular Vesicle Isolation | Size Exclusion Chromatography, Differential Ultracentrifugation Kits | EV isolation for paracrine effect studies | Critical for investigating MSC-EV therapeutic applications [26] |
| Cryopreservation Solutions | DMSO-based cryoprotectant, Programmable freezer | Long-term cell storage | Maintains viability and functionality during long-term storage |
Molecular Mechanisms and Signaling Pathways
The therapeutic effects of alternative MSCs are mediated through complex signaling pathways that regulate tissue repair, immunomodulation, and angiogenesis. The following diagram illustrates key mechanistic pathways:
Diagram 2: MSC Therapeutic Mechanism Pathways
UC-MSCs secrete higher levels of hepatocyte growth factor (HGF), basic fibroblast growth factor-2 (FGF-2), and nerve growth factor (NGF) compared to BM-MSCs and AD-MSCs [26]. HGF stimulates proliferation and exhibits anti-apoptotic and anti-fibrotic properties, while FGF-2 synergizes with VEGF to stimulate angiogenesis. MenSCs demonstrate upregulated expression of matrix metalloproteinases (MMPs) and angiogenic factors that facilitate tissue remodeling and repair [27].
The immunomodulatory properties vary between sources. AD-MSCs show significantly higher indoleamine 2,3-dioxygenase (IDO) and factor H expression, while BM-MSCs demonstrate higher CTLA-4 and IL-10 levels [29]. This suggests source-specific immunomodulatory profiles that may dictate therapeutic suitability for different conditions.
Functional Differences and Preclinical Applications
Table 3: Functional Properties and Research Applications
| Functional Attribute | UC-MSCs | PMSCs | MenSCs |
|---|---|---|---|
| Immunomodulatory Strength | Potent (low HLA-DR expression) [26] | Pronounced (DC and T-cell suppression) [3] | Moderate (low immunogenicity) [27] |
| Angiogenic Potential | High (VEGF, FGF-2 secretion) [26] | Moderate | High (uterine tissue remodeling) [28] |
| Migration Capacity | High (inflammatory homing) | Moderate | Exceptional (endometrial recruitment) |
| Key Research Applications | Cardiovascular repair, neurological disorders, GvHD [30] | Immune-mediated disorders, tissue engineering | Endometrial regeneration, ovarian restoration, Asherman's syndrome [25] [28] |
| EV Therapeutic Potential | High (similar to parent cells) [26] | Under investigation | High (wound healing, cardiac repair) [27] |
Umbilical cord, placental, and menstrual blood-derived MSCs represent scientifically valid and therapeutically promising alternatives to traditional BM and AT sources. Each source offers distinct advantages: UC-MSCs provide robust proliferation and potent immunomodulation; PMSCs offer unique immunosuppressive properties; and MenSCs deliver exceptional expansion capacity and endometrial homing capabilities [3] [27] [26].
Future research directions should focus on standardizing isolation and characterization protocols across laboratories, particularly for MenSCs and PMSCs where established standards are still evolving. The therapeutic potential of MSC-derived extracellular vesicles represents a particularly promising avenue, potentially offering similar efficacy with reduced regulatory hurdles [27] [26]. As the field advances, understanding the nuanced functional differences between these alternative MSC sources will enable researchers to select optimal cells for specific therapeutic applications, ultimately accelerating the development of effective regenerative therapies.
Mesenchymal stem cells (MSCs) represent a cornerstone of regenerative medicine, with their therapeutic potential primarily mediated through three core mechanisms: multipotent differentiation capacity, potent paracrine signaling, and immunomodulation. While bone marrow (BM) has traditionally been the primary source for MSC isolation, adipose tissue (AT) has emerged as a highly accessible and biologically distinct alternative. This technical review provides an in-depth analysis of these mechanistic pillars, presenting structured comparative data between BM-MSCs and AT-MSCs, detailed experimental protocols for functional validation, and visualizations of critical signaling pathways. The synthesis of current research underscores that the choice between MSC sources must be strategically aligned with the specific therapeutic application, whether the goal is tissue regeneration, immunomodulation, or trophic support.
Mesenchymal stem cells (MSCs), also termed mesenchymal stromal cells, are fibroblast-like, plastic-adherent cells with multipotent differentiation capacity [31] [32]. The International Society for Cellular Therapy (ISCT) has established minimal defining criteria: (1) expression of CD73, CD90, and CD105 surface markers in ≥95% of the population, combined with lack of expression (≤2%) of hematopoietic markers (CD45, CD34, CD14, CD11b, CD79α, CD19, and HLA-DR); (2) plastic adherence under standard culture conditions; and (3) ability to differentiate into osteoblasts, adipocytes, and chondroblasts in vitro [20] [32]. While initially isolated from bone marrow, MSCs are now sourced from diverse tissues, including adipose tissue, umbilical cord, and dental pulp [32]. For clinical applications, bone marrow and adipose tissue remain the most extensively studied and utilized sources, each conferring distinct biological advantages and limitations that influence their therapeutic profile [33] [20].
Core Therapeutic Mechanism 1: Multipotent Differentiation
The capacity for trilineage mesodermal differentiation is a functional hallmark of MSCs. However, significant source-dependent variations in differentiation potency directly impact their suitability for specific regenerative applications.
Table 1: Comparative Differentiation Potential of BM-MSCs vs. AT-MSCs
| Differentiation Lineage | BM-MSC Performance | AT-MSC Performance | Key Comparative Findings |
|---|---|---|---|
| Osteogenic | High | Moderate | BM-MSCs demonstrate superior calcium deposition, higher Alkaline Phosphatase (ALP) activity, and increased expression of osteogenesis-related genes (e.g., osteopontin) [33] [20]. |
| Chondrogenic | High | Moderate | BM-MSCs exhibit a greater capacity to form cartilage matrix and express chondrogenesis-related genes [33] [20]. |
| Adipogenic | Moderate | High | AT-MSCs show significantly greater potential, with more extensive lipid vesicle formation and higher expression of adipogenesis-related genes [33] [20]. |
Experimental Protocol: Trilineage Differentiation
The following standardized in vitro protocols are used to validate MSC multipotency [34].
1. Osteogenic Differentiation:
- Seeding: Plate MSCs at a density of 1.0 x 10^5 cells per well in a fibronectin-coated 6-well plate.
- Culture: Grow cells to 80-90% confluency in growth medium (e.g., MSC Growth Medium 2).
- Induction: Replace growth medium with Osteogenic Differentiation Medium (e.g., PromoCell C-28013), typically containing dexamethasone, ascorbate, and β-glycerophosphate.
- Maintenance: Differentiate for 12-14 days, changing the medium every 72 hours.
- Analysis: Fix cells and stain with Alizarin Red S to detect extracellular calcium deposits, which appear orange-red [34].
2. Adipogenic Differentiation:
- Seeding: Plate MSCs at a density of 1.0 x 10^5 cells per well.
- Culture: Grow to 80-90% confluency.
- Induction: Induce with Adipogenic Differentiation Medium (e.g., PromoCell C-28016), often containing insulin, indomethacin, and dexamethasone.
- Maintenance: Differentiate for 12-14 days, with medium changes every 3 days.
- Analysis: Fix cells and stain with Oil Red O to visualize intracellular lipid droplets, which stain bright red [34].
3. Chondrogenic Differentiation:
- Seeding: Pellet 2.0 x 10^5 MSCs in a 96-well U-bottom suspension plate.
- Spheroid Formation: Incubate for 24-48 hours to allow spontaneous spheroid (micromass) formation.
- Induction: Induce spheroids with Chondrogenic Differentiation Medium (e.g., PromoCell C-28012), typically containing TGF-β3.
- Maintenance: Differentiate for 21 days, changing medium every 3 days.
- Analysis: Fix spheroids and stain with Alcian Blue to detect sulfated proteoglycans in the cartilage matrix, which stain dark blue [34].
Diagram 1: Trilineage differentiation workflow for MSCs.
Core Therapeutic Mechanism 2: Paracrine Signaling
The primary therapeutic benefits of MSCs are now largely attributed to their potent paracrine activity, rather than direct engraftment and differentiation [31] [35]. MSCs secrete a complex mixture of bioactive factors—collectively known as the "secretome"—including growth factors, cytokines, chemokines, and extracellular vesicles, which orchestrate tissue repair by modulating local cellular responses [31] [35].
Table 2: Key Secretome Factors and Their Therapeutic Roles
| Secreted Factor Category | Example Molecules | Primary Therapeutic Functions |
|---|---|---|
| Growth Factors | HGF, bFGF, IGF-1, VEGF | Angiogenesis, anti-apoptosis, cell proliferation, and chemoattraction [33] [31]. |
| Cytokines | IL-6, IFN-γ | Immunomodulation, macrophage polarization [33] [36]. |
| Chemokines | SDF-1 | Stem cell homing and recruitment [33]. |
| Immunomodulatory Mediators | PGE2, IDO, TGF-β1 | Suppression of T-cell proliferation, induction of Tregs [32] [36]. |
The composition of the secretome is highly dynamic and influenced by the MSC tissue source and the local microenvironment. For instance, AT-MSCs secrete higher levels of bFGF, IFN-γ, and IGF-1, while BM-MSCs secrete more SDF-1 and HGF [33]. Furthermore, the secretome is not static; it is dynamically shaped by the inflammatory cues in the immediate tissue microenvironment, allowing MSCs to respond in a context-specific manner [35].
Core Therapeutic Mechanism 3: Immunomodulation
MSCs possess broad immunomodulatory properties that suppress adaptive and innate immune responses, making them potent therapeutic agents for autoimmune diseases and graft-versus-host disease (GVHD) [37] [38] [36]. This immunomodulation is mediated through direct cell-cell contact and the secretion of soluble factors.
Mechanisms of Action on Immune Cells
- T-Lymphocytes: MSCs suppress the proliferation of CD4+ and CD8+ T cells and shift the balance from a pro-inflammatory Th1 profile (IFN-γ) to an anti-inflammatory Th2 profile (IL-4) [32] [36]. A key mechanism is the promotion of Regulatory T cells (Tregs), which are induced by MSC-secreted factors like PGE2 and TGF-β, and via interactions with MSC-primed monocytes [36].
- Monocytes/Macrophages: MSCs drive the polarization of macrophages from a pro-inflammatory (M1) to an anti-inflammatory, tissue-reparative (M2) phenotype. This is achieved through the secretion of PGE2, IL-6, and IL-1RA [36]. Notably, even the phagocytosis of apoptotic or dead MSCs by monocytes can induce this anti-inflammatory polarization [38] [36].
- Dendritic Cells (DCs): MSCs inhibit the maturation and antigen-presenting capacity of DCs, reducing their ability to activate T cells [36].
- B-Lymphocytes and NK Cells: MSCs inhibit B cell proliferation and antibody production, and suppress the proliferation and cytotoxic activity of Natural Killer (NK) cells [32].
Viability-Independent Effects
Remarkably, the immunomodulatory function of MSCs is not solely dependent on cell viability. Apoptotic, metabolically inactivated, and fragmented MSCs have also demonstrated significant immunomodulatory potential, primarily through mechanisms involving phagocytosis by monocytes [38] [36]. This finding has important implications for safety and the development of cell-free therapeutic products.
Diagram 2: MSC immunomodulation mechanisms on innate and adaptive immunity.
Comparative Analysis: Bone Marrow vs. Adipose Tissue MSCs
Direct, donor-matched comparisons are essential for elucidating the intrinsic differences between BM-MSCs and AT-MSCs, minimizing donor-dependent variability [20].
Table 3: Head-to-Head Comparison of BM-MSCs and AT-MSCs
| Biological Characteristic | BM-MSCs | AT-MSCs | Research Implications |
|---|---|---|---|
| Source Availability | Limited, invasive harvest [20] | Abundant, minimally invasive harvest [20] [39] | AT-MSCs allow for larger cell yields with lower donor morbidity. |
| Proliferation Rate | Lower population doubling [33] | Significantly higher proliferative potential [33] [20] | AT-MSCs reach therapeutic cell numbers faster, advantageous for autologous therapy. |
| Immunophenotype (Typical) | CD73+, CD90+, CD105+, Stro-1+ [20] | CD73+, CD90+, CD105+, CD49d+, Stro-1low [20] | Surface marker Stro-1 and CD49d may be useful for source identification. |
| Osteogenic Potential | High [33] [20] | Moderate [33] [20] | BM-MSCs may be preferred for bone regeneration applications. |
| Chondrogenic Potential | High [33] [20] | Moderate [33] [20] | BM-MSCs may be superior for cartilage repair. |
| Adipogenic Potential | Moderate [33] [20] | High [33] [20] | AT-MSCs are the benchmark for adipogenic studies. |
| Immunomodulatory Capacity | Potent [33] | More potent in some studies [33] | AT-MSCs may offer enhanced suppression of immune responses. |
| Key Secreted Factors | Higher SDF-1, HGF [33] | Higher bFGF, IFN-γ, IGF-1 [33] | Secretome profile dictates suitability for specific paracrine applications. |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for MSC Research
| Reagent / Material | Primary Function | Example Use-Case |
|---|---|---|
| Fetal Bovine Serum (FBS) | Standard supplement for basal culture medium provides growth factors and adhesion factors [37]. | Routine expansion of MSCs. |
| Human Platelet Lysate (hPL) | Xeno-free, human-derived FBS alternative; enhances MSC proliferation and is suitable for clinical-grade expansion [33] [37]. | Clinical-scale expansion of MSCs for therapeutic applications. |
| Recombinant Human FGF-2 | Growth factor supplement that decreases population doubling time and increases expansion efficiency [37]. | Enhancing proliferation rates during in vitro culture. |
| Collagenase Type I/IV | Enzymatic digestion of tissues (e.g., adipose tissue, bone marrow) to isolate the stromal vascular fraction (SVF) containing MSCs [33] [20]. | Initial isolation of MSCs from raw tissue. |
| Flow Cytometry Antibodies | Characterization of MSC immunophenotype per ISCT criteria (CD73, CD90, CD105, CD34, CD45, etc.) [33] [20]. | Quality control and validation of MSC identity. |
| Trilineage Differentiation Kits | Pre-formulated media containing inducters for osteogenesis, adipogenesis, and chondrogenesis [34]. | Functional validation of MSC multipotency. |
| Cell Culture-grade Heparin | Anticoagulant required for culture media supplemented with hPL to prevent gel formation [33]. | Essential when using hPL-supplemented media. |
The therapeutic efficacy of MSCs is a multifaceted interplay of their multipotent differentiation capacity, dynamic paracrine signaling, and context-dependent immunomodulation. The strategic choice between BM-MSCs and AT-MSCs is not a matter of superiority, but rather of matching the biological strengths of each cell source to the specific therapeutic goal. BM-MSCs, with their robust osteogenic and chondrogenic potential, may be the source of choice for orthopedic applications. In contrast, the high proliferative yield, potent immunomodulation, and strong adipogenic capacity of AT-MSCs make them an excellent candidate for treating inflammatory conditions and for soft tissue engineering. A deep understanding of these core mechanisms, coupled with standardized functional assays and a recognition of source-specific attributes, is paramount for advancing the next generation of MSC-based therapies from the laboratory to the clinic.
From Bench to Bedside: Isolation, Expansion, and Therapeutic Applications
Standardized Protocols for Isolating and Expanding BM-MSCs and AD-MSCs
Mesenchymal stromal cells (MSCs) have emerged as powerful tools in regenerative medicine due to their self-renewal capacity, multilineage differentiation potential, and immunomodulatory properties [2]. According to the International Society for Cell & Gene Therapy (ISCT), MSCs are defined by three key criteria: adherence to plastic under standard culture conditions; expression of specific surface markers (CD73, CD90, CD105 ≥95%; lack of hematopoietic markers (CD34, CD45, CD14/CD11b, CD79α/CD19, HLA-DR ≤2%); and tri-lineage differentiation potential into osteocytes, adipocytes, and chondrocytes in vitro [2] [3]. While MSCs can be isolated from various tissues, including umbilical cord, placenta, and dental pulp, bone marrow and adipose tissue represent the most extensively studied and clinically utilized sources [9] [3]. This whitepaper provides a detailed technical guide to the standardized protocols for isolating and expanding bone marrow-derived MSCs (BM-MSCs) and adipose-derived MSCs (AD-MSCs), framing these methodologies within the broader context of MSC source selection for research and therapeutic development.
Bone Marrow-Derived MSCs (BM-MSCs): Isolation and Expansion
Isolation Techniques for BM-MSCs
Bone marrow represents the most established source of MSCs, though BM-MSCs are present at a low frequency of approximately 0.001%–0.01% of bone marrow mononuclear cells, necessitating effective isolation and expansion protocols [13]. The standard isolation approaches include:
- Bone Marrow Aspiration: Bone marrow is typically aspirated from the donor's posterior iliac crest using a Jamshidi needle into a heparin-containing syringe to prevent coagulation [40]. Aspiration volumes should be limited to approximately 2 mL per site to avoid peripheral blood dilution [40].
- Density Gradient Centrifugation: This method enriches the mononuclear cell fraction using media such as Ficoll or Percoll with densities of 1.073–1.077 g/mL [40] [9]. The automated Sepax cell separation system has demonstrated enhanced mononuclear cell recovery compared to manual Ficoll separation, improving standardization [40].
- Direct Plating Method: As an alternative to density gradient separation, the direct plating strategy separates plastic-adherent MSCs from non-adherent hematopoietic cells through adherence capability [40]. While this method often results in initial hematopoietic contamination, subsequent passaging efficiently removes non-MSC populations [40]. Recommended plating densities range from 4–22 × 10³ bone marrow mononuclear cells (BM-MNCs)/cm² [40].
Table 1: Comparison of BM-MSC Isolation Methods
| Method | Procedure | Advantages | Limitations |
|---|---|---|---|
| Density Gradient Centrifugation | Separation using Ficoll/Percoll | Reduces hematopoietic cell contamination | Requires additional equipment and processing time |
| Direct Plating | Immediate plating of BM aspirate | Simpler, fewer processing steps | Initial hematopoietic contamination |
| Whole Bone Marrow Culture | Culturing flushed marrow with fat mass [41] | Minimal disturbance to MSC niche | Specific to mouse model research |
For mouse BM-MSC isolation, a specialized protocol involves flushing bones with complete α-MEM medium and culturing the entire marrow content, including fat mass, without filtering or washing, thereby maintaining MSCs in their initial niche with minimal disturbance [41]. This method has demonstrated success across multiple mouse strains without requiring additional growth factors or cell sorting techniques [41].
Expansion and Culture of BM-MSCs
Once isolated, BM-MSCs require ex vivo expansion to achieve clinically relevant numbers. Key considerations for expansion include:
- Basal Media: Various basal media are employed, with α-MEM and DMEM being the most common [40] [13]. The critical component remains the serum supplement, which significantly influences MSC growth and functionality.
- Serum Considerations: Traditional MSC expansion relies on fetal bovine serum (FBS), though it presents significant safety concerns including risk of xenogenic immune reactions and transmission of infectious agents [40] [13]. Regulatory agencies increasingly discourage FBS use in clinical applications, driving the adoption of human alternatives like human platelet lysate (hPL), which demonstrates superior growth-promoting properties while mitigating xenogenic risks [40] [13].
- Serum-Free Media (SFM): Commercially available SFM formulations (e.g., StemMACS MSC XF, MSC NutriStem XF) support BM-MSC expansion while addressing regulatory concerns [13]. These defined media eliminate batch-to-batch variability and enhance manufacturing consistency, though they require validation for specific MSC sources and applications [13].
Table 2: BM-MSC Expansion Media Comparison
| Media Type | Examples | Advantages | Disadvantages |
|---|---|---|---|
| Fetal Bovine Serum (FBS) | Standard FBS | Well-characterized, widely used | Xenogenic risk, batch variability |
| Human Platelet Lysate (hPL) | PLTMax hPL | Human-derived, superior growth | Variable composition, requires pooling |
| Serum-Free/Xeno-Free Media | StemMACS MSC XF, MSC NutriStem XF | Defined composition, regulatory compliance | May require adaptation, cell-specific |
Research indicates that BM-MSCs expanded in SFM maintain characteristic surface marker expression, though parameters such as population doubling time, cell yield, differentiation potential, and immunosuppressive properties may vary between formulations [13]. BM-MSCs cultured in RoosterNourish (containing 1% FBS) and RoosterNourish-MSC XF typically appear spindle-shaped and elongated, while those in StemMACS MSC XF and MSC NutriStem XF present as spindle-shaped but shorter and thicker compared to control cultures [13].
Figure 1: BM-MSC Isolation and Expansion Workflow
Adipose-Derived MSCs (AD-MSCs): Isolation and Expansion
Isolation Techniques for AD-MSCs
Adipose tissue represents an abundant source of MSCs, with frequencies potentially 100–1,000 times higher than those in bone marrow [40]. The isolation process involves multiple steps:
- Tissue Harvesting: Adipose tissue is typically obtained through liposuction procedures, with smaller volumes collected via syringe aspiration or excision [40]. Tumescent liposuction involving pre-infusion of saline solutions with anesthetics and vasoconstrictors is common, though ultrasound-assisted liposuction may compromise MSC recovery and expansion capacity [40].
- Processing and Enzymatic Digestion: The lipoaspirate undergoes extensive washing to remove cellular debris, oil, blood cells, and extracellular matrix components [40]. The tissue is then digested with collagenase enzymes (typically 0.075% collagenase type I or II) at 37°C with agitation for 30-60 minutes [9].
- Stromal Vascular Fraction (SVF) Separation: Following digestion, the tissue is centrifuged to separate the mature adipocytes (floating layer) from the stromal vascular fraction (pellet) [40]. The SVF represents a heterogeneous cell mixture containing endothelial cells, pericytes, fibroblasts, and AD-MSCs [40].
- Plastic Adherence: The SVF is plated on culture vessels, allowing AD-MSCs to adhere while removing non-adherent cells through medium changes [40]. Approximately 1:1,000 cells within the SVF give rise to colony-forming units-fibroblastic (CFU-F), indicative of MSC frequency [40].
Automated closed systems have been developed for clinical-scale AD-MSC isolation, performing aspiration, washing, and SVF concentration at the patient's bedside, enhancing standardization and reducing contamination risk [40].
Expansion and Culture of AD-MSCs
AD-MSC expansion follows principles similar to BM-MSCs, with some distinct considerations:
- Culture Media: AD-MSCs proliferate effectively in standard MSC media such as DMEM or α-MEM supplemented with either FBS or human alternatives like platelet lysate [40]. Studies indicate that AD-MSCs from young, healthy donors can demonstrate rapid population doubling, potentially twice the rate observed in BM-MSCs [3].
- Morphological Characteristics: AD-MSCs typically exhibit a fibroblast-like, spindle-shaped morphology similar to BM-MSCs, though subtle differences in growth patterns and cell size may be observed [13].
- Donor Considerations: Unlike BM-MSCs, AD-MSC quantity and quality appear less affected by donor age and comorbidities such as obesity, renal failure, or vascular disease [40].
Figure 2: AD-MSC Isolation and Expansion Workflow
Comparative Analysis: BM-MSCs vs. AD-MSCs
Biological and Functional Comparisons
Understanding the relative advantages and limitations of BM-MSCs and AD-MSCs informs appropriate source selection for specific applications:
- Frequency and Accessibility: AD-MSCs exist at significantly higher frequencies in their native tissue compared to BM-MSCs (approximately 1-2% of nucleated cells in adipose tissue versus 0.001%-0.01% in bone marrow) [40] [13]. While AD-MSC harvesting is less invasive, bone marrow aspiration remains a well-tolerated procedure [16].
- Proliferation Capacity: Studies conflict regarding comparative proliferation rates, though some indicate AD-MSCs may proliferate faster than BM-MSCs, potentially reducing time to achieve target cell numbers [3] [16].
- Differentiation Potential: Both cell types demonstrate tri-lineage differentiation, though subtle lineage-specific biases exist. AD-MSCs may exhibit enhanced adipogenic propensity, while BM-MSCs often show superior osteogenic capacity [16].
- Immunomodulatory Properties: The immunomodulatory capacities of both MSC types are well-established, though direct comparative studies show conflicting results regarding their relative potency [16]. In vitro studies suggesting superior immunosuppression by AD-MSCs have not consistently translated to in vivo models [16].
- Transcriptional and Secretory Profiles: Transcriptomic and proteomic analyses reveal differences in gene expression patterns and secretory profiles between BM-MSCs and AD-MSCs, which may influence their functional applications [16].
Table 3: Functional Comparison of BM-MSCs and AD-MSCs
| Parameter | BM-MSCs | AD-MSCs |
|---|---|---|
| Tissue Frequency | 0.001%-0.01% [13] | 1-2% (of nucleated cells) [40] |
| Harvesting Procedure | Bone marrow aspiration [40] | Liposuction/aspiration [40] |
| Osteogenic Potential | Strong [16] | Moderate [16] |
| Adipogenic Potential | Moderate [16] | Strong [16] |
| Clinical Trial Experience | Extensive (10 approved therapies) [3] | Limited (2 approved therapies) [3] |
| Regulatory Status | Well-established pathway [42] | Less established [16] |
Technical and Regulatory Considerations
From a technical and regulatory perspective, several factors distinguish these MSC sources:
- Manufacturing Standardization: BM-MSC manufacturing benefits from longer development history and established international standards (ISO/TS 24651:2022) [3]. Multiple clinical trials have successfully manufactured BM-MSCs according to Good Manufacturing Practice (GMP) standards for bone regeneration and other applications [42].
- Product Characterization: BM-MSCs typically demonstrate strong expression of standard MSC markers (CD44, CD90) with negative expression of hematopoietic markers (CD45, CD31) [41]. Both cell types must meet ISCT criteria for surface marker expression, though subtle differences in marker intensity or additional markers may occur.
- Donor Variability: BM-MSC quantity and quality are influenced by donor age, with younger donors generally providing more robust cells [3]. AD-MSCs appear less affected by donor age and comorbidities, potentially offering more consistent cell products across diverse donor populations [40].
- Therapeutic Applications: Sixteen MSC therapies have been approved worldwide: ten derived from bone marrow, three from umbilical cord, two from adipose tissue, and one from umbilical cord blood [3]. This disparity reflects both historical development and regulatory familiarity with BM-MSCs.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for MSC Isolation and Expansion
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Basal Media | α-MEM, DMEM [40] [13] | Nutrient support for cell growth and maintenance |
| Serum Supplements | Fetal Bovine Serum (FBS) [13] | Traditional source of growth factors and attachment factors |
| Human Supplements | Human Platelet Lysate (hPL) [40] [13] | Xeno-free alternative to FBS for clinical applications |
| Serum-Free Media | StemMACS MSC XF, MSC NutriStem XF [13] | Chemically defined media for standardized, regulatory-compliant expansion |
| Digestion Enzymes | Collagenase Type I/II [9] | Tissue dissociation for MSC isolation from adipose tissue |
| Density Gradient Media | Ficoll-Paque, Percoll [40] [9] | Separation of mononuclear cells from bone marrow aspirates |
| Antibiotics | Penicillin-Streptomycin-Neomycin (PSN) [41] | Prevention of microbial contamination in primary cultures |
| Trypsin/EDTA | 0.25% Trypsin-EDTA [41] | Cell detachment during passaging |
| Characterization Antibodies | CD73, CD90, CD105, CD34, CD45, HLA-DR [2] [3] | Flow cytometry analysis to confirm MSC phenotype |
The selection between BM-MSCs and AD-MSCs represents a critical decision point in research and therapeutic development. BM-MSCs benefit from extensive characterization, established protocols, and regulatory precedent, while AD-MSCs offer higher initial cell yields and less invasive harvesting. Both cell types present unique advantages that may suit different applications—BM-MSCs for conditions requiring robust osteogenesis or leveraging their homing to bone marrow niches, and AD-MSCs for applications benefiting from their abundant availability and potentially enhanced angiogenic properties. As the field advances, standardized protocols across both cell sources will be essential for generating comparable, reproducible data and advancing MSC-based therapies through regulatory pathways to clinical application. Future directions will likely focus on further optimization of xeno-free culture systems, development of potency assays predictive of therapeutic efficacy, and standardization of production processes across different MSC sources.
The expansion of mesenchymal stem cells (MSCs) for regenerative medicine has historically relied on fetal bovine serum (FBS) as a primary culture supplement. However, FBS presents significant risks, including batch-to-batch variability, potential xenogeneic immunogenic reactions, and ethical concerns related to animal welfare [33] [43] [44]. Human platelet lysate (hPL) has emerged as a viable, xenogeneic-free alternative derived from human platelet concentrates. Rich in growth factors like PDGF, VEGF, EGF, and FGF, hPL supports robust MSC proliferation while aligning with clinical safety standards by eliminating zoonotic pathogen risks [45] [46] [47]. This whitepaper details the transition to hPL for clinical-grade MSC culture, emphasizing bone marrow (BM-) and adipose tissue (AT-) derived MSCs.
hPL in MSC Biology: Proliferation, Differentiation, and Functional Enhancement
Impact on MSC Proliferation and Phenotype
hPL significantly enhances MSC proliferation rates compared to FBS. Studies report a ~2-fold increase in cell yield and shorter population doubling time in hPL-supplemented media [48]. For instance, umbilical cord tissue-derived MSCs cultured in hPL exhibited a population doubling time of 20.95 hours versus 22.25 hours in FBS [48]. Both BM-MSCs and AT-MSCs retained typical fibroblast-like morphology and surface marker expression (CD73+, CD90+, CD105+, CD34-, CD45-) in hPL, confirming adherence to ISCT criteria [33] [20] [48].
Differentiation Potential and Tissue-Specific Advantages
hPL not only maintains but can enhance MSC differentiation capacity:
- Osteogenesis: BM-MSCs demonstrated superior osteogenic potential in hPL, with higher alkaline phosphatase activity and calcium deposition [33] [20].
- Chondrogenesis: BM-MSCs also showed increased chondrogenic capacity compared to AT-MSCs in hPL [33].
- Adipogenesis: AT-MSCs exhibited greater lipid vesicle formation and adipogenic gene expression [20].
- Endothelial Differentiation: hPL enhanced endothelial-specific gene expression in human amniotic fluid MSCs, promoting tube formation on Matrigel [45].
Table 1: Comparative Analysis of BM-MSCs vs. AT-MSCs in hPL Supplemented Culture
| Parameter | BM-MSCs in hPL | AT-MSCs in hPL |
|---|---|---|
| Proliferation Rate | Moderate | Higher [33] |
| Osteogenic Potential | High [33] [20] | Moderate [33] [20] |
| Chondrogenic Potential | High [33] | Moderate [33] |
| Adipogenic Potential | Moderate [20] | High [20] |
| Immunomodulatory Effects | Moderate | More potent [33] |
| Secreted Factors | Higher SDF-1, HGF [33] | Higher bFGF, IFN-γ, IGF-1 [33] |
Functional and Therapeutic Benefits
- Cardiac Repair: BM-MSCs pre-treated with hPL (PMSCs) transplanted into infarcted rat hearts improved systolic/diastolic pressure, reduced infarct size, and enhanced angiogenesis [49].
- Immunomodulation: hPL-supplemented MSCs showed enhanced suppression of T-cell proliferation in mixed lymphocyte reactions, critical for allogeneic therapies [48].
- Bone Remodeling: hPL supported both osteogenic differentiation of MSCs and osteoclastic differentiation of monocytes in co-culture, enabling balanced bone remodeling models [44].
Standardizing hPL Manufacturing: Protocols and Quality Control
hPL Production Workflow
Protocol: hPL Preparation via Freeze-Thaw Method
- Platelet Concentrate Sourcing: Use expired human platelet concentrates (PCs) pooled from multiple donors to minimize donor variation [46] [47].
- Freeze-Thaw Cycles: Subject PCs to 3–4 cycles at -80°C (freezing) and 37°C (thawing) to lyse platelets and release growth factors [46] [48].
- Centrifugation & Filtration: Centrifuge at 4,000 × g for 10–15 min to remove debris; filter supernatant through 0.22 µm filters for sterilization [46] [47].
- Storage: Aliquot and store at -80°C; lyophilization (L-HPL) can enhance stability without altering growth factor content [46].
Quality Control and Standardization Metrics
To ensure batch-to-batch consistency, assess:
- Growth Factor Concentrations: PDGF-AA/BB, TGF-β1, VEGF, FGF2 via Luminex assays [47].
- Biochemical Parameters: pH (7.2–7.6), osmolality (280–320 mOsm/kg), total protein content [46] [47].
- Functional Assays: MSC doubling time, tri-lineage differentiation, and immunophenotype validation [47] [48].
Table 2: Key Quality Attributes for hPL Batches
| Parameter | Target Range | Method |
|---|---|---|
| Total Protein | 30–50 mg/mL | Spectrophotometry [46] |
| pH | 7.2–7.6 | Electrode meter [47] |
| Osmolality | 280–320 mOsm/kg | Blood gas analyzer [47] |
| PDGF-AB | 50–150 ng/mL | Multiplex immunoassay [47] |
| VEGF | 0.5–2 ng/mL | Multiplex immunoassay [47] |
| Microbial Safety | Sterile | BacT/ALERT or PCR [47] |
Diagram 1: hPL Manufacturing and MSC Culture Workflow (Max Width: 760px)
The Scientist’s Toolkit: Essential Reagents for hPL-Based MSC Culture
Table 3: Key Research Reagent Solutions for hPL-MSC Culture
| Reagent/Catalog | Function | Application Note |
|---|---|---|
| Platelet Concentrates | Source of growth factors (PDGF, VEGF, TGF-β) | Pool 4–8 donor units to minimize variability [46] [47] |
| Heparin | Prevents fibrin clot formation in hPL supplements | Use at 2–5 IU/mL in culture media [44] |
| Lymphoprep | Density gradient medium for mononuclear cell isolation | Critical for BM-MSC and PBMC-derived co-cultures [44] |
| Collagenase Type I/IV | Enzymatic digestion of adipose tissue or umbilical cord | Yields stromal vascular fraction for AT-MSC isolation [20] [48] |
| Trypsin-EDTA | Passaging adherent MSCs | Standardized concentration (0.05%) for cell detachment [20] |
| Luminex Assay Kits | Quantify growth factors (PDGF, VEGF, TGF-β) in hPL batches | Ensures batch-to-batch consistency [47] |
| Matrigel | Assess endothelial differentiation and tube formation | Validate angiogenic potential of hPL-cultured MSCs [45] |
Experimental Protocols: Key Methodologies for hPL Translation
Protocol: Direct Comparison of BM-MSCs vs. AT-MSCs in hPL
Base Media: Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 5–10% hPL and 2–4 IU/mL heparin [33].
- Cell Isolation:
- Expansion: Culture at 37°C/5% CO₂; passage at 80–90% confluence using trypsin-EDTA.
- Analysis:
Protocol: Functional Validation in Disease Models
Cardiac Infarction Model [49]:
- Pre-treat BM-MSCs with 10% hPL for 72 hours to generate PMSCs.
- Inject PMSCs (or HPL alone) into infarcted rat myocardium.
- Assess hemodynamics (LVSP, ±dp/dt max) and histology (infarct size, CD31+ angiogenesis) after 4 weeks.
Osteoclast-Osteoblast Co-Culture [44]:
- Culture CD14+ monocytes with BM-MSCs in 2.5–10% hPL.
- Differentiate with M-CSF/RANKL for osteoclasts and ascorbate/β-glycerophosphate for osteoblasts.
- Measure resorption pits (calcium phosphate coatings) and OPG/RANKL ratios.
Diagram 2: Experimental Design for MSC Source Comparison in hPL (Max Width: 760px)
The transition from FBS to hPL represents a critical advancement in clinical-grade MSC manufacturing. hPL offers a standardized, ethically compliant, and functionally superior supplement that enhances MSC proliferation while maintaining differentiation capacity and therapeutic efficacy. Key considerations for adoption include:
- Source Selection: BM-MSCs for orthopedic applications due to superior osteochondral potential; AT-MSCs for immunomodulation and volume-sensitive therapies [33] [20].
- Standardization: Implement multi-donor pooling and rigorous QC of growth factors to minimize variability [47].
- Regulatory Alignment: Use pathogen-inactivated platelet concentrates and defined protocols compliant with Good Manufacturing Practices (GMP) [46] [47].
By integrating hPL into MSC culture systems, researchers can accelerate the development of safe, effective cell therapies for regenerative medicine.
Mesenchymal stem cells (MSCs) represent a cornerstone of regenerative medicine due to their remarkable capacity for self-renewal and multilineage differentiation potential. These non-hematopoietic, multipotent stem cells can be isolated from various tissues, including bone marrow, adipose tissue, umbilical cord, and dental pulp, and are defined by specific criteria established by the International Society for Cellular Therapy (ISCT) [50] [2]. According to these guidelines, MSCs must be adherent to plastic under standard culture conditions; express the surface markers CD105, CD73, and CD90 while lacking expression of hematopoietic markers (CD45, CD34, CD14 or CD11b, CD79α or CD19, and HLA-DR); and possess the capacity to differentiate into osteoblasts, chondrocytes, and adipocytes in vitro [51] [2]. The balance between these differentiation pathways is crucial for maintaining tissue homeostasis, and its dysregulation contributes to various disease states, including osteoporosis, where adipogenic differentiation becomes favored over osteogenesis [51] [52].
The therapeutic potential of MSCs extends beyond their differentiation capabilities to include immunomodulatory functions and paracrine effects through the secretion of bioactive molecules [2]. However, harnessing this potential requires a deep understanding of the specific molecular mechanisms and signaling pathways that govern lineage commitment. This technical guide provides a comprehensive overview of the current methodologies, molecular regulators, and experimental protocols for directing MSCs toward osteogenic, chondrogenic, and adipogenic lineages, with a particular focus on MSCs derived from bone marrow and adipose tissue within the context of regenerative medicine and disease modeling.
Molecular Mechanisms Governing MSC Differentiation
The fate determination of MSCs is precisely regulated by a complex interplay of signaling pathways, transcription factors, and epigenetic modifications. Understanding these molecular mechanisms is fundamental to controlling differentiation processes for therapeutic applications.
Key Signaling Pathways
Wnt/β-Catenin Signaling: The Wnt signaling pathway plays a dual role in MSC differentiation, with context-dependent effects. Canonical Wnt signaling, mediated through β-catenin stabilization, promotes osteogenic commitment by inhibiting adipogenesis through suppression of peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer-binding protein α (C/EBPα) [50]. However, activated Wnt/β-catenin signaling can inhibit terminal osteoblast differentiation in osteogenic conditions [50]. Non-canonical Wnt members, particularly Wnt5a, have been shown to stimulate osteoblast differentiation through autocrine loops in human MSCs [50].
BMP/TGF-β Signaling: Bone morphogenetic proteins (BMPs), members of the TGF-β superfamily, signal primarily through Smad proteins (Smad1/5/8) and play crucial roles in osteogenic differentiation [50]. BMP-2, -4, and -7 can induce adipocyte commitment in MSCs by activating PPARγ expression [53]. Interestingly, TGF-β signaling has dual effects on adipogenesis, potentially related to MSC origin and heterogeneity [53]. In osteogenesis, TGF-β signaling generally inhibits osteoblastic maturation, and its inhibition has been shown to enhance osteogenic differentiation in adipose-derived stromal cells (ASC), fibroblasts (FB), and dental pulp stromal cells (DSC) [54].
Rho GTPase Signaling: The Rho family of GTPases, including RhoA, Rac1, and Cdc42, regulates actin cytoskeleton reorganization, which mechanically influences MSC lineage commitment. Inhibition of RhoA or its downstream effector ROCK promotes adipogenesis by reducing cellular tension, while activation favors osteogenesis [53]. Actin cytoskeleton reorganization from aligned filaments to a disorganized meshwork is characteristic of adipogenic differentiation [53].
Master Transcription Factors and Epigenetic Regulation
Lineage-specific transcription factors serve as master regulators of MSC differentiation. Runt-related transcription factor 2 (Runx2) directs osteogenic differentiation [53], while Sox9 regulates chondrogenesis [53]. For adipogenesis, PPARγ and C/EBPα form a core transcriptional cascade that promotes adipocyte maturation [53]. Recent research has identified Matrix Gla Protein (MGP) as a positive regulator of adipogenic differentiation in MSCs through the Ca2+/CaMKII/RIP140/FABP3 axis, with implications for osteoporosis [52].
MicroRNAs and other non-coding RNAs provide an additional layer of regulation by fine-tuning the expression of these transcription factors and signaling components [50] [51]. Furthermore, epigenetic modifications, including DNA methylation and histone acetylation, dynamically regulate differentiation by modulating chromatin accessibility at key gene promoters [53] [51].
Table 1: Master Transcription Factors in MSC Differentiation
| Lineage | Key Transcription Factors | Primary Functions |
|---|---|---|
| Osteogenic | Runx2, Osterix | Master regulators of osteoblast differentiation; activate bone matrix protein genes |
| Chondrogenic | Sox9, SOX5, SOX6 | Essential for chondrocyte differentiation and cartilage matrix production |
| Adipogenic | PPARγ, C/EBPα, C/EBPβ | Core adipogenic regulators; control lipogenic gene expression |
Osteogenic Differentiation
Molecular Mechanisms
Osteogenic differentiation of MSCs is a sequential process culminating in the formation of mineralized bone matrix. This process is primarily regulated by the Wnt/β-catenin and BMP signaling pathways [50]. BMPs, particularly BMP-2, -4, and -7, activate Smad1/5/8 phosphorylation, which in turn induces the expression of Runx2, the master transcription factor for osteoblastogenesis [50] [54]. Runx2 activates osteogenic marker genes including those encoding type I collagen, osteocalcin, and alkaline phosphatase (ALP) [50]. The canonical Wnt pathway enhances osteogenic commitment by inhibiting adipogenic transcription factors PPARγ and C/EBPα [50]. Additionally, non-canonical Wnts such as Wnt5a stimulate osteoblast differentiation through autocrine mechanisms [50].
Vitamin D3, ascorbic acid, and β-glycerophosphate are essential components of osteogenic induction media. Vitamin D3 promotes mineralization through genomic actions via vitamin D response elements (VDREs) in target genes like osteocalcin in human MSCs [50]. Ascorbic acid is crucial for collagen synthesis and matrix maturation, while β-glycerophosphate provides a phosphate source for ALP-mediated mineralization, leading to apatite crystal deposition [50].
Standard Induction Protocol
Materials:
- Basal medium: α-MEM or DMEM-low glucose
- Fetal bovine serum (FBS): 10%
- Antibiotic-Antimycotic: 1%
- Ascorbic acid: 50-100 μM
- β-glycerophosphate: 10 mM
- Dexamethasone: 100 nM (optional, enhances differentiation but not specific to osteogenesis) [50]
Procedure:
- Culture MSCs to 80% confluence in growth medium.
- Replace with osteogenic induction medium.
- Change medium every 3-4 days for 21-28 days.
- Monitor differentiation through ALP activity (early marker, days 7-14) and calcium deposition (late marker, from day 14 onward).
Analysis Methods:
- ALP staining or activity assay (days 7-14)
- Alizarin Red S staining for calcium deposition (days 21-28)
- Immunostaining or PCR for osteocalcin, osteopontin, Runx2
- Von Kossa staining for mineralization
Factors Influencing Osteogenic Potential
The osteogenic capacity of MSCs varies significantly depending on tissue origin and donor characteristics. Dental pulp stromal cells (DSCs) demonstrate superior osteogenic potential compared to adipose-derived stromal cells (ASCs) and fibroblasts (FBs), with BMP-2 supplementation further enhancing differentiation in DSCs but inhibiting it in ASCs [54]. This difference correlates with increased expression of BMP-2 receptors ALK-3 and ALK-6 in DSCs [54].
Replicative senescence also markedly affects osteogenic potential. While ASCs maintain consistent osteogenic capacity through later passages (P10), DSCs show a dramatic reduction in differentiation potential with senescence [54]. TGF-β signaling inhibition enhances osteogenic differentiation in early-passage cells, but this benefit diminishes in senescent cells [54].
Table 2: Osteogenic Potential Across MSC Sources
| MSC Source | Osteogenic Potential | Response to BMP-2 | Effect of Senescence |
|---|---|---|---|
| Bone Marrow | High (gold standard) | Variable enhancement | Moderate decrease |
| Adipose Tissue | Moderate | Inhibitory | Minimal decrease |
| Dental Pulp | Very High | Significantly enhanced | Severe decrease |
| Umbilical Cord | Moderate | Not well documented | Minimal decrease |
Figure 1: Osteogenic Signaling Pathways. Key pathways including canonical Wnt and BMP signaling regulate osteogenic differentiation through transcriptional activation of osteogenic genes.
Chondrogenic Differentiation
Molecular Mechanisms and Challenges
Chondrogenic differentiation of MSCs presents unique challenges due to the complex structure and composition of native hyaline cartilage. Unlike osteogenesis and adipogenesis, chondrogenesis typically requires three-dimensional culture systems to achieve proper cellular condensation and matrix production [55]. The process is primarily regulated by transforming growth factor-beta (TGF-β) superfamily members, particularly TGF-β1, TGF-β3, and BMPs, which activate Smad2/3 and Smad1/5/8 signaling pathways, respectively [55] [56].
A significant limitation in MSC-based chondrogenesis is the tendency toward hypertrophic differentiation, mimicking endochondral ossification rather than forming stable articular cartilage [55]. MSC-derived chondrocytes typically express hypertrophy markers including type X collagen, alkaline phosphatase, and matrix metalloproteinase 13 (MMP13), ultimately leading to matrix mineralization and vascular invasion in vivo [55]. This default pathway reflects the developmental origin of many MSC populations and presents a major obstacle for clinical cartilage repair strategies.
The transcription factor Sox9 is the master regulator of chondrogenesis, cooperating with SOX5 and SOX6 to activate cartilage-specific genes including type II collagen and aggrecan [55] [56]. Recent approaches to enhance chondrogenesis and suppress hypertrophy include biomimetic scaffolds incorporating cartilage-specific matrix components such as hyaluronan (HA) and chondroitin sulfate (CS), which can modulate signaling through CD44 and TGF-β pathways [56].
Standard Induction Protocol
Materials:
- Basal medium: DMEM-high glucose
- ITS+ supplement (Insulin-Transferrin-Selenium): 1%
- Antibiotic-Antimycotic: 1%
- Sodium pyruvate: 1 mM
- Proline: 40 μg/mL
- Ascorbic acid: 50 μg/mL
- Dexamethasone: 100 nM
- TGF-β1 or TGF-β3: 10 ng/mL
Procedure:
- Harvest MSCs and create micromass cultures (centrifuge 2.5×10^5 cells in 15 mL conical tube) or seed into 3D scaffolds.
- Replace with chondrogenic induction medium.
- Change medium every 3-4 days for 21-35 days.
- Maintain in culture for up to 6 weeks for matrix maturation.
Analysis Methods:
- Histological staining: Alcian blue or Safranin O for proteoglycans
- Immunohistochemistry: Type II collagen, aggrecan
- PCR: Sox9, type II collagen, aggrecan, type X collagen (hypertrophy marker)
- Biochemical assays: Glycosaminoglycan (GAG) quantification
Enhancing Chondrogenic Outcomes
The chondrogenic niche significantly influences differentiation outcomes. Biomaterial scaffolds mimicking native cartilage extracellular matrix can enhance chondrogenesis while suppressing hypertrophy. Polyelectrolyte multilayer coatings combining native hyaluronan with type I collagen have shown promise in promoting cartilage-specific differentiation through CD44 receptor binding and activation of ERK/Sox9 and TGF-β-p38 signaling pathways [56].
Additionally, chondroitin sulfate in scaffold materials has been demonstrated to delay or prevent hypertrophic progression, though the precise mechanism remains unclear [55]. Physical properties of the culture environment, including substrate stiffness, also critically influence chondrogenic commitment, with softer matrices generally favoring chondrogenesis over osteogenesis [56].
Figure 2: Chondrogenic Signaling Pathways. TGF-β signaling and CD44 activation through hyaluronan binding promote chondrogenic differentiation through Sox9 activation.
Adipogenic Differentiation
Molecular Mechanisms
Adipogenic differentiation of MSCs is a complex, multi-stage process involving commitment of multipotent MSCs to preadipocytes and subsequent terminal differentiation into mature adipocytes [53]. This process is coordinated by a cascade of transcription factors, with PPARγ and C/EBPα serving as the master regulators that activate adipogenic gene expression programs [53] [52]. The early phase of differentiation involves expression of C/EBPβ and C/EBPδ, which in turn activate PPARγ and C/EBPα, establishing a positive feedback loop that maintains the differentiated state [53].
Several signaling pathways converge to regulate adipogenesis. BMP signaling, particularly through BMP2 and BMP4, promotes adipocyte commitment by inducing PPARγ expression [53]. BMP7 has been shown to specifically stimulate brown adipocyte differentiation [53]. TGF-β/Smad signaling generally suppresses adipogenesis by inhibiting C/EBPα and PPARγ expression, though the effects may vary depending on MSC source and specific experimental conditions [53]. Actin cytoskeleton remodeling and Rho GTPase signaling also play crucial roles, with reduced actin tension and inhibition of RhoA/ROCK signaling promoting adipogenic commitment [53].
Recent research has identified Matrix Gla Protein (MGP) as a significant positive regulator of MSC adipogenesis through a novel mechanism involving increased intracellular Ca2+ levels, enhanced CaMKII phosphorylation, and subsequent RIP140 protein degradation, leading to increased transcription of FABP3, a fatty acid transport protein [52]. This MGP/Ca2+/CaMKII/RIP140/FABP3 axis provides new insights into the molecular control of adipogenic differentiation and its relationship with bone metabolic disorders.
Standard Induction Protocol
Materials:
- Basal medium: DMEM-high glucose
- Fetal bovine serum (FBS): 10%
- Antibiotic-Antimycotic: 1%
- Isobutylmethylxanthine (IBMX): 0.5 mM
- Dexamethasone: 1 μM
- Indomethacin: 200 μM
- Insulin: 10 μg/mL
Procedure:
- Culture MSCs to 100% confluence (growth arrest).
- Replace with adipogenic induction medium.
- After 3 days, change to adipogenic maintenance medium (DMEM + 10% FBS + Insulin).
- Cycle between induction (2-3 days) and maintenance (2-3 days) media for 2-3 cycles.
- Continue culture in maintenance medium for 10-15 days total.
Analysis Methods:
- Oil Red O staining for lipid droplets
- PCR: PPARγ, C/EBPα, FABP4, adiponectin
- Immunostaining: Perilipin 1
- Triglyceride quantification assay
Disease Connections and Metabolic Regulation
Adipogenic differentiation of MSCs is not only fundamental to normal adipose tissue homeostasis but also plays a critical role in various disease states. In osteoporosis, an imbalance between adipogenic and osteogenic differentiation leads to excessive bone marrow adipose tissue formation at the expense of bone formation [51] [52]. MGP expression is significantly upregulated in osteoporosis and shows a strong negative correlation with bone mineral density, highlighting its potential as a therapeutic target [52].
Metabolic regulation is integral to adipogenic differentiation, with insulin enhancing glucose uptake for triglyceride synthesis [53]. IBMX increases intracellular cAMP levels through phosphodiesterase inhibition, while dexamethasone activates glucocorticoid receptors to induce C/EBPδ expression [53]. Indomethacin promotes adipogenesis through PPARγ activation independent of its cyclooxygenase inhibitory activity [53].
Table 3: Adipogenic Induction Media Components and Functions
| Component | Concentration | Primary Function |
|---|---|---|
| IBMX | 0.5 mM | Phosphodiesterase inhibitor; increases cAMP; stimulates C/EBPβ |
| Dexamethasone | 1 μM | Glucocorticoid receptor agonist; induces C/EBPδ expression |
| Indomethacin | 200 μM | PPARγ activator; promotes adipogenesis |
| Insulin | 10 μg/mL | Enhances glucose uptake; supports triglyceride synthesis |
Figure 3: Adipogenic Signaling Pathways. Multiple pathways including early transcriptional activation and MGP signaling converge on PPARγ and C/EBPα to drive adipogenic differentiation.
Experimental Considerations and Technical Challenges
MSC Source Selection
The tissue origin of MSCs significantly impacts their differentiation potential and biological properties. Bone marrow-derived MSCs (BM-MSCs) remain the most extensively characterized population, but adipose tissue-derived MSCs (ASCs) offer advantages including higher yield and less invasive isolation procedures [57] [2]. Neonatal tissue-derived MSCs, such as those from umbilical cord (UC-MSCs), typically exhibit superior proliferation capacity and longer life spans compared to adult tissue-derived MSCs [57].
Recent research has identified functional differences even between MSC populations isolated from different compartments within the same anatomical site. Bone marrow adipose tissue-derived MSCs (BMAT-MSCs) demonstrate significantly higher adipogenic and osteogenic differentiation potential compared to conventional BM-MSCs from the same cavity [58]. These differences extend to pathological conditions, with Fanconi anemia and acute myeloid leukemia samples showing distinct differentiation impairments and molecular profiles [58].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for MSC Differentiation Studies
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Induction Cocktails | Osteogenic: Ascorbic acid, β-glycerophosphate, DexamethasoneChondrogenic: TGF-β1/3, BMP-6, ITS+ PremixAdipogenic: IBMX, Dexamethasone, Indomethacin, Insulin | Direct lineage-specific differentiation |
| Signaling Modulators | SB431542 (TGF-β inhibitor)Dorsomorphin (BMP inhibitor)Recombinant BMP-2 (osteogenic enhancer)CHIR99021 (Wnt activator) | Pathway-specific manipulation to study mechanisms |
| Detection Reagents | Alizarin Red S (calcium deposition)Oil Red O (lipid droplets)Alcian Blue (proteoglycans)Antibodies against: Osteocalcin, Type II Collagen, Perilipin | Assessment of differentiation efficiency and matrix production |
| Cell Culture Systems | 3D aggregate culture systemsChondrogenic micromass cultureBiomimetic scaffolds (HA, Col I, CS) | Provide appropriate microenvironment for differentiation |
Technical Challenges and Optimization Strategies
Several technical challenges persist in MSC differentiation studies. The tendency of MSC-derived chondrocytes to undergo hypertrophy remains a significant obstacle for cartilage repair applications [55]. Optimization strategies include careful temporal control of TGF-β exposure, incorporation of chondroitin sulfate in scaffold materials, and use of specific hyaluronan formulations to promote stable chondrogenesis [55] [56].
Replicative senescence represents another critical consideration, particularly for clinical applications requiring extensive cell expansion. The impact of senescence varies by MSC source, with DSC showing dramatic reductions in osteogenic potential at later passages while ASC maintain more consistent differentiation capacity [54]. Donor age and health status also significantly influence MSC function, with MSCs from osteoporosis patients showing elevated adipogenic potential [52].
Standardization of differentiation protocols remains challenging due to variations in MSC isolation methods, culture conditions, and serum batches. Researchers should implement rigorous quality control measures including comprehensive characterization of differentiation outcomes using multiple complementary assessment methods to ensure reproducible and meaningful results.
The directed differentiation of MSCs into osteogenic, chondrogenic, and adipogenic lineages represents a powerful tool for both basic research and clinical applications in regenerative medicine. While standardized protocols exist for each lineage, the efficiency and stability of differentiation are influenced by numerous factors including MSC source, donor characteristics, culture conditions, and specific induction methods. A comprehensive understanding of the molecular mechanisms underlying lineage commitment, particularly the key signaling pathways and transcriptional networks, enables researchers to optimize differentiation protocols for specific applications. Future advances in this field will likely focus on enhancing the stability of chondrogenic differentiation, developing more precise temporal control of signaling pathways, and creating biomimetic culture systems that better recapitulate the native stem cell niche. The continued refinement of MSC differentiation protocols holds significant promise for tissue engineering, disease modeling, and cell-based therapies for a wide range of degenerative conditions.
Mesenchymal stem/stromal cells (MSCs) have emerged as next-generation drug delivery vehicles for cancer therapeutics and other applications, representing a paradigm shift from conventional delivery systems [59]. These multipotent stromal cells possess innate tumor-homing capabilities, allowing them to migrate toward tumor tissues and inflammatory microenvironments following administration [59] [60]. This unique tropism, combined with their ability to be loaded with diverse therapeutic payloads, positions MSCs as promising living vectors for targeted therapy. MSCs can be isolated from multiple tissue sources, including bone marrow, adipose tissue, umbilical cord, and placenta, with each source offering distinct advantages and limitations [61] [20] [60]. The foundational principle underlying MSC-based delivery systems is their capacity to carry cytotoxic loads while demonstrating remarkable resistance to toxic effects, enabling them to transport and release therapeutics at disease sites [60]. This technical guide examines current engineering strategies for enhancing MSC therapeutic cargo delivery, framed within the context of comparative MSC source biology and its implications for drug delivery optimization.
Biological Foundations of MSCs for Drug Delivery
MSC Heterogeneity and Source Selection
The selection of MSC source material significantly influences cellular properties, expansion potential, and ultimately therapeutic performance. MSCs from different anatomical niches exhibit substantial functional heterogeneity that must be considered when designing drug delivery systems. Bone marrow-derived MSCs (BM-MSCs) represent the most extensively characterized population but present challenges including painful harvesting procedures and low yield frequency (approximately 1 in 3.4 × 10^4 cells) [61] [20]. Adipose-derived stem cells (ASCs) offer a compelling alternative with significantly higher yields—approximately 5,000 stem cells per gram of adipose tissue compared to 100–1,000 cells/mL from bone marrow aspirate [61]. Donor-matched comparisons reveal that while ASCs demonstrate superior proliferation rates and adipogenic capacity, BM-MSCs exhibit enhanced osteogenic and chondrogenic differentiation potential [20]. This tissue-specific differentiation capacity varies significantly among donors, highlighting the importance of donor selection and source matching for specific therapeutic applications [20].
Table 1: Comparative Analysis of Primary MSC Sources for Drug Delivery Applications
| Source | Harvesting Procedure | Cell Yield | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Bone Marrow | Invasive, painful aspiration [20] | Low (100-1,000 cells/mL) [61] | Gold standard, well-characterized [20] | Donor age-dependent decline [60] |
| Adipose Tissue | Minimally invasive (liposuction) [20] | High (~5,000 cells/g) [61] | Abundant tissue, rapid expansion [61] [20] | Reduced osteogenic vs. BM-MSCs [20] |
| Placenta (hAMSCs) | Non-invasive, ethically uncontroversial [60] | Very high [60] | Immunoprivileged, unique homing [60] | Less established protocols [60] |
Tumor Homing Mechanisms and Administration Routes
The tumor-homing capacity of MSCs constitutes their principal advantage as targeted delivery vehicles. This homing ability is mediated by complex chemokine-cytokine interactions between MSC receptors and ligands expressed within the tumor microenvironment [59]. The inflammatory signals secreted by tumors create a chemotactic gradient that guides MSC migration following administration. The route of MSC administration significantly influences homing efficiency and biodistribution. Intravenous injection remains the most common approach but poses challenges including pulmonary first-pass effect, where a significant proportion of cells may be trapped in lung capillaries [59]. Intra-arterial delivery offers more direct access to certain tumor sites, while local implantation provides high regional concentration but limited dissemination [59]. Optimizing administration protocols represents an active area of investigation to maximize tumor delivery efficiency while minimizing off-target distribution.
Engineering Strategies for MSC Cargo Loading
Small Molecule Drug Loading
The loading of MSCs with small molecule chemotherapeutics has demonstrated promising results in preclinical models. A pivotal study using human amniotic mesenchymal stromal cells (hAMSCs) demonstrated successful paclitaxel (PTX) loading and release kinetics sufficient to inhibit pancreatic tumor cell (CFPAC-1) proliferation in vitro [60]. Notably, hAMSCs exhibited remarkable resistance to PTX cytotoxicity, surviving concentrations that would be lethal to most cell types [60]. This intrinsic resistance mechanism is partially attributed to the expression of P-glycoprotein (P-gp), a multidrug efflux transporter that may protect MSCs from chemotherapeutic toxicity while simultaneously influencing drug loading and release profiles [60]. Similar approaches have successfully loaded doxorubicin and other chemotherapeutics into MSCs from various tissue sources, confirming the broad applicability of this strategy [62].
Genetic Engineering for Therapeutic Protein Expression
Genetic modification of MSCs enables sustained production and secretion of therapeutic proteins, including cytokines, growth factor antagonists, and pro-apoptotic proteins [59] [60]. This approach typically involves viral vector-mediated transduction to introduce genes encoding therapeutic proteins under the control of constitutive or inducible promoters. Engineered MSCs can thus serve as local bioreactors within the tumor microenvironment, continuously producing anti-neoplastic agents. While promising in animal models, genetic manipulation of MSCs for clinical application requires careful risk-benefit analysis due to potential insertional mutagenesis and immune reactions to viral vectors [60]. Non-viral approaches including electroporation and nucleofection offer alternatives with improved safety profiles, though typically with reduced transfection efficiency [62].
Extracellular Vesicle Engineering
MSC-derived extracellular vesicles (MSC-EVs) represent a promising cell-free alternative that leverages the inherent therapeutic properties of MSCs while overcoming limitations associated with whole-cell therapies [63]. EVs offer advantages including reduced immunogenicity, enhanced stability, and the ability to cross biological barriers [63] [62]. Engineering strategies for EV cargo loading can be categorized into pre-isolation (endogenous) and post-isolation (exogenous) methods:
Table 2: Extracellular Vesicle Cargo Loading Strategies
| Method | Mechanism | Ideal Cargo Types | Key Considerations |
|---|---|---|---|
| Co-incubation | Passive diffusion across EV membrane [62] | Lipophilic drugs (curcumin, paclitaxel, doxorubicin) [62] | Preserves EV integrity; limited to small lipophilic molecules [62] |
| Electroporation | Electrical impulses create transient membrane pores [62] | Nucleic acids (siRNA, miRNA, DNA) [62] | Risk of cargo aggregation; may compromise membrane integrity [62] |
| Sonication | Ultrasonic waves disrupt EV membrane [62] | Hydrophilic drugs, proteins, nucleic acids [62] | High loading efficiency; potential damage to surface proteins [62] |
| Membrane Permeabilization | Chemical reagents (saponin) create pores [62] | Oligonucleotides, proteins [62] | Milder on EV structure; risk of residual contaminant [62] |
Recent advances in EV engineering focus on modulating protein-lipid interactions to enhance cargo loading. Engineering proteins to associate with lipid raft domains through specific transmembrane domains or lipidation motifs significantly improves EV loading efficiency [64]. Bioinformatic analysis reveals that natural EV proteins possess distinctive features including longer transmembrane domains and specific post-translational modifications such as palmitoylation and prenylation, providing design principles for engineered constructs [64].
Experimental Protocols for MSC Drug Loading
Protocol: Small Molecule Drug Loading (Paclitaxel Example)
This protocol adapts methodology from hAMSC studies for general MSC drug loading applications [60]:
Reagents and Materials:
- MSCs (bone marrow, adipose, or placental origin)
- Paclitaxel stock solution (5 mg/mL in DMSO)
- Complete culture medium (DMEM with 10% FBS)
- Phosphate-buffered saline (PBS)
- Cell culture vessels (T-flasks, multiwell plates)
- Centrifuge tubes
Procedure:
- Culture MSCs to 70-80% confluence in appropriate growth medium.
- Prepare working PTX solutions by diluting stock in culture medium to desired concentrations (typically 1-1000 ng/mL).
- Replace existing medium with PTX-containing medium.
- Incubate cells for 24 hours under standard culture conditions (37°C, 5% CO2).
- Remove drug-containing medium and wash cells thoroughly with PBS to remove surface-adherent drug.
- Harvest drug-loaded MSCs using standard trypsinization techniques.
- Validate drug loading through HPLC-MS or functional cytotoxicity assays.
Technical Notes:
- Optimal loading concentration should be determined empirically for each MSC source and drug combination.
- Include viability assessment post-loading (trypan blue exclusion or MTT assay).
- For in vivo applications, ensure thorough washing to prevent carry-over of unincorporated drug.
Protocol: EV Loading via Electroporation
This protocol describes nucleic acid loading into MSC-EVs for RNA-based therapeutics [62]:
Reagents and Materials:
- Isolated MSC-EVs (via ultracentrifugation or size-exclusion chromatography)
- Nucleic acid cargo (siRNA, miRNA)
- Electroporation buffer (typically containing sucrose and MgCl2)
- Electroporation cuvettes (4 mm gap)
- Electroporator system
- DNA/RNA quantification reagents
Procedure:
- Isolate EVs from MSC-conditioned media using preferred method (ultracentrifugation recommended).
- Characterize EV quantity (protein assay) and size (nanoparticle tracking analysis).
- Mix 100 μg EVs with 5-10 μg nucleic acid in electroporation buffer.
- Transfer mixture to pre-chilled electroporation cuvette.
- Apply electrical pulses (typically 500 V, 125 μF, 5 pulses for 500 μL volume).
- Incubate on ice for 30 minutes to allow membrane recovery.
- Remove unencapsulated nucleic acid through ultrafiltration or size-exclusion chromatography.
- Quantify loading efficiency using fluorescently-labeled nucleic acids or RT-qPCR.
Technical Notes:
- Addition of EDTA to electroporation buffer can minimize nucleic acid aggregation [62].
- Include controls for non-specific nucleic acid attachment.
- Validate functional delivery in recipient cells.
EV Loading Workflow: Schematic representation of the electroporation-based loading protocol for encapsulating nucleic acids in extracellular vesicles.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for MSC Drug Delivery Studies
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| MSC Isolation | Collagenase Type I [61] [20], Dispase [60], Density gradient media | Tissue dissociation and MSC isolation | Concentration and digestion time vary by tissue source [61] |
| Cell Culture | DMEM/F12 medium [20], Fetal Bovine Serum [20], Human platelet lysate [65] | MSC expansion and maintenance | Serum-free alternatives reduce batch variability [65] |
| Characterization | CD73, CD90, CD105 antibodies [61] [20] [60], CD34, CD45 antibodies [61] [20] | Flow cytometry immunophenotyping | ≥95% positive for MSC markers; ≤2% positive for hematopoietic markers [20] |
| Drug Loading | Paclitaxel [60], Doxorubicin [62], Synthetic nucleic acids [62] | Therapeutic cargo loading | Cytotoxicity assessment essential post-loading [60] |
| EV Isolation | Ultracentrifugation system [64], Size-exclusion columns [66], PEG-based precipitation [62] | Extracellular vesicle purification | Method choice affects yield, purity, and functionality [66] |
Visualization of Key Signaling Pathways in MSC Therapy
The therapeutic effects of MSCs and their secretomes are mediated through modulation of key signaling pathways in recipient cells. In hepatocellular carcinoma models, MSC-derived factors influence multiple oncogenic pathways including NF-κB, Wnt/β-catenin, Notch1, Stat3, and TGF-β [67]. The activation state of these pathways determines whether MSCs exert tumor-suppressive or tumor-promoting effects, highlighting the context-dependent nature of MSC therapy [67].
MSC Signaling Pathways: Key molecular pathways modulated by MSC-derived factors that influence tumor behavior in a context-dependent manner.
Clinical Translation Challenges and Future Perspectives
The translation of MSC-based drug delivery systems from preclinical models to clinical application faces several significant challenges. Interdonor heterogeneity of MSCs represents a major obstacle to consistent therapeutic outcomes, as biological properties including proliferation and differentiation capacity vary significantly among donors [59] [20]. Additional hurdles include optimizing tumor-homing efficiency, controlling drug release kinetics, and ensuring consistent manufacturing quality under Good Manufacturing Practice (GMP) standards [59] [65]. As of July 2025, ClinicalTrials.gov reports 1163 registered clinical trials focused on stem cell therapy, primarily addressing central nervous system disorders, autoimmune diseases, GVHD, and osteogenesis [65]. Future directions include the development of enhanced homing strategies through chemokine receptor engineering, improved loading techniques utilizing novel materials science approaches, and combination therapies that leverage MSCs to deliver multiple therapeutic modalities simultaneously [59] [64]. The continued refinement of MSC engineering strategies promises to unlock the full potential of these remarkable cellular vehicles for targeted therapeutic delivery.
Mesenchymal stem cells (MSCs) have emerged as a powerful tool in regenerative medicine due to their multipotent differentiation potential, immunomodulatory properties, and low immunogenicity. Initially identified in bone marrow by Friedenstein and colleagues in the 1970s, MSCs are now known to reside in various tissues, including adipose tissue, umbilical cord, and placental tissue [2] [68]. The therapeutic application of MSCs has expanded dramatically from foundational biological concepts to clinically approved treatments for specific conditions. The International Society for Cellular Therapy (ISCT) established minimal criteria for defining MSCs, including plastic adherence, specific surface marker expression (CD73, CD90, CD105), and trilineage differentiation potential (osteogenic, chondrogenic, adipogenic) [3] [2]. This framework has enabled standardization across research and clinical applications, facilitating the transition from laboratory discovery to clinical implementation.
The clinical translation of MSC therapies represents a paradigm shift in treating conditions with high unmet medical needs. Rather than relying solely on cell replacement through differentiation, evidence indicates that MSCs exert their therapeutic effects primarily through sophisticated paracrine signaling and immunomodulation [68]. These mechanisms include the secretion of bioactive molecules such as growth factors, cytokines, and extracellular vesicles that modulate local cellular environments, promote tissue repair, stimulate angiogenesis, and exert anti-inflammatory effects [2] [68]. More recently, mitochondrial transfer has been identified as a novel therapeutic mechanism where MSCs donate mitochondria to injured cells, restoring cellular bioenergetics in conditions such as acute respiratory distress syndrome and myocardial ischemia [68]. This comprehensive review examines the current clinical translation landscape of MSC therapies, focusing on approved applications for graft-versus-host disease (GvHD), Crohn's fistula, and osteoarthritis, while providing technical insights into the experimental frameworks underpinning these advances.
Approved MSC-Based Therapies and Clinical Applications
The global regulatory landscape for MSC therapies has evolved significantly, with numerous products receiving marketing authorization for specific clinical indications. To date, sixteen MSC therapies have been approved worldwide: ten derived from bone marrow, three from umbilical cord, two from adipose tissue, and one from umbilical cord blood [3]. These approvals represent milestones in the field, providing novel treatment options for conditions previously managed with limited success using conventional approaches. The market for MSC therapies is projected to grow from USD 3.87 billion in 2025 to USD 13.49 billion by 2035, reflecting increasing clinical adoption and development of new applications [69].
Table 1: Globally Approved MSC-Based Therapies and Their Clinical Applications
| Product Name/Type | Tissue Source | Indication | Mechanism of Action | Approval Status |
|---|---|---|---|---|
| Remestemcel-L | Bone Marrow | Pediatric steroid-refractory acute GvHD | Immunomodulation via T-cell suppression and anti-inflammatory macrophage polarization | Approved (Phase III trial showed 70.4% overall response at day 28) [68] |
| RYONCIL | Mesenchymal stromal cells | Pediatric steroid-refractory acute GvHD | Immune modulation through paracrine signaling and direct cell contact | Approved 2024 [70] |
| MSC-based products | Adipose Tissue | Complex anal fistulas in Crohn's disease | Immunomodulation and tissue repair via paracrine effects | Approved (two products) [3] |
| MSC-based products | Umbilical Cord | Osteoarthritis, other indications | Cartilage regeneration and anti-inflammatory effects | Approved (three products) [3] |
| MC0518 (investigational) | Mesenchymal stromal cells | Steroid-refractory acute GvHD | Immune-modulating capabilities to reduce inflammatory response | Phase III trials (Overall response rate at Day 28 primary endpoint) [70] |
The clinical efficacy of MSC therapies is particularly evident in immune-mediated conditions. For graft-versus-host disease (GvHD), a serious complication of allogeneic hematopoietic stem cell transplantation, MSC products have demonstrated significant clinical benefits. In a pivotal Phase III trial of Remestemcel-L, an MSC product derived from bone marrow, infusions markedly alleviated symptoms in pediatric patients with steroid-refractory acute GvHD, with an overall response rate of 70.4% at day 28 and durable benefit [68]. Similarly, RYONCIL received approval in 2024 for pediatric steroid-refractory acute GvHD, representing the continuing expansion of this therapeutic class [70]. Additional MSC products, such as MC0518, are currently in late-stage clinical development for steroid-refractory acute GvHD, further validating this approach [70].
For complex perianal fistulas in Crohn's disease, approved MSC therapies derived from adipose tissue have demonstrated capacity to modulate the local inflammatory environment and promote tissue repair through paracrine mechanisms [3]. The transition from traditional surgical approaches to cell-based therapies represents a significant advancement in managing this challenging condition. Similarly, for osteoarthritis, approved umbilical cord-derived MSC products have shown promise in promoting cartilage regeneration and reducing joint inflammation, addressing a substantial unmet need in this common degenerative condition [3].
Experimental Models and Methodologies in MSC Research
In Vitro Characterization and Differentiation Protocols
Standardized experimental protocols are essential for evaluating MSC potency and differentiation capacity prior to clinical application. The ISCT characterization standards require specific methodologies to confirm MSC identity and functional potential [3] [2].
Immunophenotypic Characterization by Flow Cytometry: MSC surface marker expression must demonstrate ≥95% positivity for CD105, CD73, and CD90, and ≤2% expression of hematopoietic markers (CD45, CD34, CD14/CD11b, CD79α/CD19, HLA-DR) [3]. Sample processing involves harvesting cells using 0.25% Trypsin/EDTA, staining with directly labeled antibodies in PBS with BSA and NaN₃ (PBN) with 2% AB serum, and analysis by flow cytometry [71].
Trilineage Differentiation Potential Assessment: The multipotency of MSCs is validated through specific differentiation protocols:
Adipogenic Differentiation: Passage 3 BM-MSCs are cultured in adipogenic differentiation medium consisting of DMEM-LG, 10% FBS, 1 μM dexamethasone, 60 μM indomethacin, 500 μM isobutylmethylxanthine, and 5 μg/mL insulin [71]. Differentiation is evaluated after 14-21 days using Oil Red O staining to visualize lipid droplets and RT-qPCR analysis of adipogenic markers PPARG and SCD [71].
Osteogenic Differentiation: Cells are induced in DMEM-LG supplemented with 10% FBS, 100 nM dexamethasone, 10 mM beta-glycerophosphate, and 0.2 mM L-ascorbic acid [71]. After 21-28 days, differentiation is assessed by Alizarin Red S staining for calcium deposition, quantitative calcium assay using the Quantichrom Calcium assay kit, and RT-qPCR for osteogenic markers ALPL and RUNX2 [71].
Chondrogenic Differentiation: A pellet culture system is employed where 2 × 10⁵ cells are transferred to 15 mL polypropylene tubes and centrifuged to form pellets [71]. Chondrogenic induction uses DMEM-HG with 1 mM sodium pyruvate, 100 nM dexamethasone, 50 μg/mL L-ascorbic acid, 10 ng/mL TGF-β3, and 1X ITS supplement [71]. Chondrogenesis is evaluated by RT-qPCR for cartilage-specific markers SOX9 and COL2.
In Vivo Disease Models for Efficacy Evaluation
Animal models provide critical preclinical data on MSC therapeutic efficacy and mechanisms of action. For GvHD research, humanized mouse models generated by transplanting human peripheral blood mononuclear cells (PBMCs) into immunodeficient mice recapitulate key aspects of human disease pathophysiology [70]. MSC efficacy is evaluated by monitoring survival rates, clinical GvHD scores, histopathological analysis of target organs (skin, liver, gastrointestinal tract), and quantification of human immune cell populations by flow cytometry [70].
For Crohn's fistula investigation, rodent models of colitis-induced fistulizing disease are utilized. The therapeutic efficacy of MSCs is assessed through histopathological evaluation of fistula tract closure, immunohistochemical analysis of inflammatory cell infiltration (CD3+ T cells, F4/80+ macrophages), and measurement of inflammatory cytokines (TNF-α, IL-6, IL-1β) in tissue homogenates [3].
In osteoarthritis research, surgical destabilization of the medial meniscus (DMM) or chemical induction models in rodents are employed. MSC treatment outcomes are evaluated through gait analysis, histopathological scoring of cartilage damage (OARSI criteria), immunohistochemistry for cartilage matrix components (collagen type II, aggrecan), and subchondral bone remodeling analysis by micro-CT [3].
Molecular Mechanisms and Signaling Pathways
The therapeutic effects of MSCs are mediated through sophisticated molecular mechanisms that vary depending on tissue source and disease context. Understanding these pathways is essential for optimizing clinical efficacy and developing potency assays.
Immunomodulatory Pathways in GvHD
In GvHD, MSCs exert immunomodulation through direct cell-cell contact and paracrine factor secretion. Key mechanisms include inhibition of T-cell proliferation through secretion of prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), and programmed death-ligand 1 (PD-L1) [68]. MSCs also guide macrophage polarization by converting pro-inflammatory M1 macrophages into anti-inflammatory M2 phenotypes through signaling molecules like interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) [68]. This shift plays a critical role in tempering overactive immune responses in GvHD.
Tissue Repair Mechanisms in Crohn's Fistula and Osteoarthritis
For Crohn's fistula, MSCs promote healing through multiple coordinated mechanisms. The secretion of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) promotes angiogenesis around fistula tracts, improving perfusion to injured areas [68]. Hepatocyte growth factor (HGF) contributes to antifibrotic effects by limiting collagen accumulation, while insulin-like growth factor 1 (IGF-1) and stromal-derived factor-1 (SDF-1) inhibit cell death and preserve tissue structure [68].
In osteoarthritis, MSCs mediate cartilage protection through different pathways. MSCs secrete extracellular vesicles containing regulatory miRNAs that modulate chondrocyte metabolism and inhibit inflammatory signaling [3] [2]. They also produce trophic factors that stimulate resident progenitor cell proliferation and differentiation, facilitating cartilage matrix synthesis and inhibiting matrix-degrading enzymes [3].
Mitochondrial Transfer Mechanism
A novel therapeutic mechanism identified more recently involves the direct transfer of mitochondria from MSCs to injured cells. Through the development of tunneling nanotubes—slender, dynamic membrane structures—MSCs can deliver healthy mitochondria directly to damaged cells, thereby restoring cellular energy production in compromised tissues [68]. This mechanism has shown significant potential in conditions characterized by mitochondrial dysfunction and is believed to contribute to the therapeutic effects observed in various inflammatory and degenerative conditions [68].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Standardized reagents and experimental systems are critical for generating reproducible, translatable MSC research data. The following table outlines essential tools and their applications in MSC characterization and potency assessment.
Table 2: Essential Research Reagent Solutions for MSC Investigations
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Surface Marker Antibodies | CD105, CD73, CD90, CD45, CD34, CD14, HLA-DR | Flow cytometric immunophenotyping per ISCT standards | ≥95% positivity for CD73, CD90, CD105; ≤2% for hematopoietic markers [3] [2] |
| Differentiation Kits | Adipogenic: Dexamethasone, indomethacin, isobutylmethylxanthine, insulin | Trilineage differentiation potential assessment | Oil Red O staining for lipid droplets; PPARG, SCD gene expression [71] |
| Osteogenic: Dexamethasone, beta-glycerophosphate, L-ascorbic acid | Osteogenic differentiation validation | Alizarin Red S staining; calcium quantification; ALPL, RUNX2 markers [71] | |
| Chondrogenic: TGF-β3, ITS supplement, sodium pyruvate | Chondrogenic capacity evaluation | Pellet culture system; SOX9, COL2 gene expression [71] | |
| Cell Culture Media | DMEM-LG/MCDB-201 mixture, Fetal Bovine Serum, Penicillin/Streptomycin | MSC expansion and maintenance | Serum batches must be pre-tested for MSC growth support; antibiotic-free culture for therapeutics [71] |
| Stress Induction Reagents | H₂O₂, tunicamycin, thapsigargin | Oxidative and ER stress challenge models | H₂DCFDA assay for ROS detection; XBP1, ATF4, CHOP for ER stress markers [71] |
| Cytokine Analysis | ELISA/Luminex for PGE2, IDO, TGF-β, IL-10 | Immunomodulatory potency assessment | Functional correlate of MSC therapeutic potential [68] |
The clinical translation landscape for MSC therapies has progressed substantially, with approved products demonstrating efficacy for specific conditions like GvHD, Crohn's fistula, and osteoarthritis. The continued evolution of this field depends on rigorous preclinical research using standardized experimental methodologies and a deep understanding of the molecular mechanisms involved. As research advances, optimizing MSC sources—including bone marrow, adipose tissue, and umbilical cord—for specific clinical applications will be essential. Future directions include enhancing MSC potency through preconditioning strategies, developing improved delivery systems, and establishing more predictive potency assays to ensure consistent clinical outcomes. The integration of mechanistic insights with robust manufacturing and quality control will further accelerate the translation of MSC therapies from bench to bedside, expanding treatment options for patients with inflammatory and degenerative diseases.
Navigating Challenges and Enhancing MSC Potency for Clinical Success
Mesenchymal stem cells (MSCs) from bone marrow and adipose tissue represent a cornerstone of regenerative medicine research, with demonstrated potential in treating orthopedic, neurological, and inflammatory conditions [72] [73]. Despite promising preclinical results and over 1,500 clinical trials investigating MSC therapies, their widespread clinical translation and commercial success face three fundamental bottlenecks: significant donor-related variability, the inevitable onset of cellular senescence during expansion, and limitations in scalable manufacturing processes [74] [15] [75]. This whitepaper provides an in-depth technical analysis of these interconnected challenges, synthesizing current research and presenting standardized experimental approaches to advance the field toward more robust and reproducible MSC-based therapeutics.
Donor Variability
Donor variability introduces profound inconsistencies in MSC proliferation capacity, differentiation potential, and secretory profile, directly impacting the efficacy and predictability of cell-based products [15] [73].
Impact of Donor Health Status
A 2025 study directly compared adipose-derived MSCs (AT-MSCs) from healthy donors and those with Type 2 Diabetes (T2D), revealing both unexpected functional capacities and limitations [15].
Table 1: Characterization of AT-MSCs from Healthy vs. Type 2 Diabetic Donors
| Parameter | AT-MSCs (Healthy Donors) | AT-MSCs (T2D Donors) |
|---|---|---|
| Morphology & Viability | Normal, adherent, >90% viability | No significant differences observed |
| Surface Marker Profile | CD73+/CD90+/CD105+/CD34-/CD45- | CD73+/CD90+/CD105+/CD34-/CD45- |
| Proliferation Rate | Standard population doubling time | No significant differences observed |
| Oxidative Stress (8OHdG) | Baseline levels | No significant differences observed |
| Senescent Cells | Baseline levels | No significant differences observed |
| Chondrogenic Potential | Standard capacity | Enhanced differentiation capacity |
| Adipogenic Potential | Standard capacity | Reduced differentiation capacity |
| Osteogenic Potential | Standard capacity | Comparable to healthy donors |
| Pro-angiogenic Secretome | Promotes HUVEC tube formation | Significantly improved HUVEC tube formation |
The study found that under standard culture conditions, AT-MSCs from T2D donors were functionally competent, with enhanced chondrogenic and pro-angiogenic potential compared to those from healthy donors [15]. This suggests that autologous MSCs from diabetic patients remain viable for developing cell-based products for specific applications like osteoarthritis or myocardial infarction [15].
Experimental Protocol: Assessing Donor-Driven Functional Variation
Objective: To evaluate the impact of donor health status on the trilineage differentiation and paracrine pro-angiogenic potential of AT-MSCs.
Methods:
- Cell Culture: Culture cryopreserved, primary passage AT-MSCs from healthy (n≥3) and T2D (n≥3) donors in standard growth medium (αMEM supplemented with 5% human platelet lysate (hPL), 2 IU/ml heparin, and 1% penicillin-streptomycin) [15].
- Trilineage Differentiation: Upon reaching 80-90% confluence, induce differentiation using commercial kits.
- Adipogenic/Osteogenic: Seed cells at 2.1 x 10^4 cells/cm². Use StemPro Adipogenesis or Osteogenesis Differentiation Kit, with medium changes every 3-4 days for 14-21 days. Fix and stain with Oil Red O (adipocytes) or Alizarin Red S (osteocytes) [15].
- Chondrogenic: Pellet 2.5 x 10^5 cells in a conical tube. Use StemPro Chondrogenesis Differentiation Kit, with medium changes every 3-4 days for 21-28 days. Analyze pellets via histology (Safranin O staining) [15].
- Pro-angiogenic Assay:
- Collect conditioned medium (CM) from AT-MSC cultures after 48 hours.
- Seed Human Umbilical Vein Endothelial Cells (HUVECs) on a Matrigel-coated surface and incubate with the CM.
- After 6-18 hours, image the formed capillary-like structures and quantify total tube length, number of nodes, and meshes per field using image analysis software (e.g., ImageJ) [15].
Senescence
Cellular senescence during in vitro expansion leads to diminished proliferative capacity, reduced multipotency, and the adoption of a pro-inflammatory secretory phenotype (SASP), severely compromising therapeutic efficacy [75].
Molecular Regulation of MSC Stemness
The stemness of MSCs—their capacity for self-renewal and differentiation—is finely regulated by a network of intrinsic genetic and epigenetic factors. Key transcriptional regulators include [75]:
- Twist1 and Twist2: These basic helix-loop-helix transcription factors are highly expressed in early-passage MSCs. Their overexpression enhances proliferation and adipogenesis while inhibiting osteogenesis. Twist1 maintains stemness by recruiting EZH2 to silence senescence genes p14 and p16 via H3K27me3 methylation [75].
- OCT4: The OCT4A isoform promotes cell cycle progression and inhibits senescence by upregulating DNMT1, which suppresses cyclin-dependent kinase inhibitors p21 and p16 [75].
- SOX2: Expression decreases with serial passaging and in senescent MSCs. SOX2 knockdown is associated with increased p16 and p21 expression, directly linking it to senescence avoidance [75].
The following diagram illustrates the core transcriptional network that maintains MSC stemness and counters senescence.
Experimental Protocol: Inducing and Quantifying Replicative Senescence
Objective: To model and assess MSC senescence resulting from serial passaging.
Methods:
- Serial Passaging: Culture BM-MSCs or AD-MSCs in standard conditions. Serially passage cells at a standardized seeding density (e.g., 1 x 10^4 cells/cm²) until proliferation significantly slows. Use passages 2-4 as "early" and passages 8-12 as "late" populations for comparison [75] [73].
- Senescence-Associated β-Galactosidase (SA-β-Gal) Staining:
- Wash cells in PBS and fix with 2% formaldehyde/0.2% glutaraldehyde for 5 minutes.
- Incubate cells with X-Gal staining solution (1 mg/mL X-Gal, 40 mM citric acid/Na phosphate buffer pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl₂) at 37°C overnight in a dry incubator without CO₂.
- Count the percentage of blue-stained cells in brightfield microscopy. A >30% positive rate is indicative of a senescent culture [75].
- qPCR Analysis of Senescence Markers:
- Extract total RNA using Trizol reagent and synthesize cDNA.
- Perform quantitative PCR using primers for key senescence-associated genes:
- CDKN2A/p16 and CDKN1A/p21 (cell cycle arrest)
- IL-6 and IL-8 (SASP components)
- Normalize expression levels to a housekeeping gene (e.g., GAPDH) and calculate fold-change relative to early-passage cells [75].
Scalable Manufacturing
Transitioning from 2D flask-based culture to controlled, scalable biomanufacturing platforms is essential for producing clinically relevant doses of MSCs and their therapeutic derivatives, such as extracellular vesicles (EVs) [74] [76].
Bioreactor Platforms for Scalable Production
Advanced bioreactor systems address the volume and process control limitations of traditional culture methods.
Table 2: Scalable Biomanufacturing Platforms for MSCs and MSC-EVs
| Platform | Process Description | Reported Yield | Key Advantages |
|---|---|---|---|
| Fixed-Bed Bioreactor | Integrated, automated system for continuous cell expansion and EV harvesting [74]. | ~1.2 × 10¹³ EV particles per day [74]. | GMP-compatible, continuous perfusion, minimal shear stress [74]. |
| Suspension Bioreactor (Microcarriers) | 3D culture of induced MSCs (iMSCs) on microcarriers in suspension [74]. | >5 × 10⁸ iMSCs per batch after 20 days [74]. | High cell density, scalable from process development to commercial scale [74]. |
| 3D Hydrogel Microenvironment | Encapsulation of MSCs in tunable 3D hydrogels to modulate cell function and EV production [77]. | Under investigation (Focus of NSF-funded research) [77]. | Recapitulates native microenvironment, modulates EV cargo and yield [77]. |
The Promise of iPSC-Derived MSCs (iMSCs)
To combat donor variability and finite expansion capacity, induced pluripotent stem cell-derived MSCs (iMSCs) offer a renewable and standardized cell source [74] [78]. iMSCs are generated by differentiating iPSCs in vitro, enabling the creation of master cell banks for unlimited, consistent starting material [78]. A 2025 study established a scalable platform using extended pluripotent stem cell (EPSC)-derived iMSCs, demonstrating robust expansion in bioreactors and production of EVs with therapeutic efficacy matching primary MSC-EVs in a model of pulmonary fibrosis [74].
The following workflow outlines the integrated biomanufacturing process for iMSC-derived extracellular vesicles.
Experimental Protocol: EV Production in a Fixed-Bed Bioreactor
Objective: To produce and isolate extracellular vesicles from iMSCs using a scalable fixed-bed bioreactor system.
Methods:
- Bioreactor Setup and Inoculation: Seed EPSC-induced iMSCs into a fixed-bed bioreactor system. Use serum-free medium to avoid contaminating serum-derived EVs. Initiate continuous perfusion to supply nutrients and remove waste products [74].
- EV Harvesting and Conditioning: Continuously harvest conditioned medium from the bioreactor over a defined production period (e.g., several days). Store the collected medium at 4°C for short-term processing [74].
- EV Isolation and Purification:
- Clarification: Centrifuge conditioned medium at 2,000 × g for 30 minutes to remove cells and large debris.
- Concentration: Use tangential flow filtration (TFF) with a 100-500 kDa molecular weight cutoff to concentrate the clarified medium.
- Purification: Further purify the EV concentrate using size-exclusion chromatography (SEC) to separate EVs from soluble proteins and other non-vesicular contaminants [74] [76].
- EV Characterization:
- Nanoparticle Tracking Analysis (NTA): Determine particle size distribution and concentration.
- Transmission Electron Microscopy (TEM): Confirm cup-shaped morphology.
- Western Blot: Analyze for positive EV markers (CD63, CD81, TSG101) and negative markers (e.g., calnexin) [74].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for MSC Research
| Reagent / Kit | Function / Application | Example Use-Case |
|---|---|---|
| Human Platelet Lysate (hPL) | Serum-free, xeno-free supplement for MSC culture medium; enhances proliferation and maintains genotype/phenotype [15]. | Standard growth medium for clinical-grade expansion of AT-MSCs and BM-MSCs [15]. |
| StemPro Differentiation Kits | Defined, ready-to-use media for inducing adipogenic, osteogenic, and chondrogenic differentiation [15]. | Standardized assessment of MSC trilineage differentiation potential per ISCT criteria [15]. |
| TrypLE Select Enzyme | Animal-origin-free, recombinant protease for cell dissociation; gentler on cells than trypsin, improving post-digestion viability [15]. | Passaging adherent MSC cultures while maintaining high cell viability and surface marker integrity [15]. |
| EPSC Induction Medium | Specialized cytokine/small molecule cocktail to reprogram iPSCs or establish extended pluripotent stem cells [74]. | Generation of EPSCs as a starting source for scalable iMSC differentiation [74]. |
| Trizol Reagent | Monophasic solution of phenol and guanidine isothiocyanate for simultaneous dissociation of biological material and inhibition of RNases [74]. | RNA extraction for downstream qPCR analysis of senescence or lineage-specific genes [74] [75]. |
The future of MSC-based therapeutics relies on systematically overcoming the intertwined challenges of donor variability, senescence, and manufacturing scalability. Strategies such as employing iMSCs from standardized banks, modulating key stemness regulators like Twist1 and SOX2 to delay senescence, and adopting integrated bioreactor systems for EV production represent the most promising paths forward [74] [75] [78]. A holistic approach that combines novel biological insights with advanced engineering principles is essential to transform mesenchymal stem cells from a research tool into reliable and effective clinical medicines.
Mesenchymal stem cells (MSCs) have emerged as highly promising candidates in regenerative medicine and immunomodulation due to their self-renewal capacity, multilineage differentiation potential, and unique immunomodulatory properties [2]. These nonhematopoietic, multipotent stem cells can be isolated from various tissues, including bone marrow, adipose tissue, and umbilical cord, making them attractive for therapeutic applications [2] [3]. However, a significant challenge has limited their clinical potential: MSCs exposed to the harsh inflammatory environment of damaged tissue after intravenous transplantation often exhibit reduced survival and weakened therapeutic function [79]. This limitation has prompted researchers to develop preconditioning strategies that "prime" MSCs in vitro to better withstand in vivo challenges and enhance their therapeutic efficacy [80].
Preconditioning involves exposing MSCs to controlled stressful conditions in vitro to activate adaptive responses that improve their survival, paracrine activity, and immunomodulatory functions after transplantation [80] [81]. Among various approaches, hypoxia and inflammatory cytokine preconditioning have demonstrated particularly promising results across multiple preclinical models [79] [80] [82]. These strategies aim to mimic the injury-induced microenvironment that MSCs encounter after administration, effectively preparing them for the challenges of the in vivo environment while boosting their therapeutic properties [79]. This technical guide explores the molecular mechanisms, methodological protocols, and clinical applications of these preconditioning approaches within the broader context of MSC research.
Preconditioning Strategies and Their Molecular Mechanisms
Hypoxia Preconditioning
Hypoxia preconditioning typically involves culturing MSCs at oxygen concentrations significantly lower than atmospheric levels (often 1-5% O₂ compared to the standard 21% O₂) for periods ranging from 24 to 72 hours [79] [82]. This approach mimics the oxygen tension in damaged tissues and activates crucial adaptive cellular responses, primarily through the stabilization of hypoxia-inducible factor-1α (HIF-1α) [82]. HIF-1α serves as a master regulator of the cellular response to low oxygen, leading to increased expression of genes involved in cell survival, metabolism, and angiogenesis [82].
The molecular mechanisms underlying hypoxia preconditioning include:
- Enhanced secretion of trophic factors including vascular endothelial growth factor (VEGF), which promotes angiogenesis and tissue repair [82]
- Activation of the Nrf2/ARE pathway, a critical antioxidant response pathway that protects cells from oxidative stress [82]
- Metabolic reprogramming toward glycolysis, which supports cell survival in low-oxygen environments [82]
- Upregulation of cell survival genes that enhance resistance to apoptosis [79]
Studies using hypoxia mimetics like cobalt chloride (CoCl₂) and deferoxamine (DFO) have confirmed that these compounds can replicate many benefits of physiological hypoxia, providing a more convenient experimental alternative to hypoxic chambers [82].
Inflammatory Cytokine Preconditioning
Inflammatory cytokine preconditioning involves exposing MSCs to pro-inflammatory cytokines such as interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), or combinations thereof [79] [80] [83]. This approach "licenses" or activates the immunomodulatory capacities of MSCs by mimicking the inflammatory microenvironment they would encounter at sites of tissue damage [80].
The molecular mechanisms of cytokine preconditioning include:
- Upregulation of immunomodulatory enzymes notably indoleamine 2,3-dioxygenase (IDO), which catalyzes the degradation of tryptophan to kynurenines, suppressing T-cell proliferation and function [80]
- Enhanced production of soluble mediators including prostaglandin E2 (PGE2), transforming growth factor-beta (TGF-β), hepatocyte growth factor (HGF), and interleukin-10 (IL-10) [80] [84]
- Increased expression of immunomodulatory surface molecules such as programmed cell death-1 ligands (PDL-1) and human leukocyte antigen-G (HLA-G) [80] [84]
- Activation of key signaling pathways including JAK-STAT and NF-κB pathways [80]
Interestingly, preconditioning with specific cytokines can also reduce donor-to-donor variability in MSC immunomodulatory potential, potentially addressing a significant challenge in clinical applications [81].
Combined Hypoxia and Inflammatory Preconditioning
Emerging evidence suggests that combining hypoxia with inflammatory cytokine preconditioning may have synergistic effects [79]. This approach more comprehensively mimics the in vivo microenvironment of damaged tissue, where both low oxygen tension and inflammatory mediators are present simultaneously [79]. Studies using this combined approach have demonstrated enhanced immunomodulatory effects without damaging fundamental MSC biological characteristics [79].
Table 1: Quantitative Effects of Different Preconditioning Strategies on MSC Properties
| Preconditioning Method | Key Molecular Changes | Functional Outcomes | Representative Studies |
|---|---|---|---|
| Hypoxia (2% O₂) | 5-10 fold increase in VEGF; Activation of Nrf2/ARE pathway | Enhanced angiogenesis; Improved cell survival under oxidative stress; Increased neuroprotective effects | [79] [82] |
| IFN-γ (10-50 ng/mL) | 10-50 fold increase in IDO expression; Upregulation of PDL-1 | Potent inhibition of T-cell proliferation; Reduced NK cell cytotoxicity; Enhanced suppression of Th17 cells | [80] [81] |
| TNF-α + IL-1β | 27-fold increase in IL-6; 720-fold increase in CCL-20; 2-7 fold increase in TSG-6 | Enhanced macrophage polarization to M2 phenotype; Reduced angiogenic potential; Improved anti-inflammatory effects | [83] |
| Combined Hypoxia + Cytokines | Significant decrease in coagulation-related tissue factors; Increased immune gene expression | Enhanced immunosuppression of PBMC and NK cell proliferation; Retention of mitochondrial function | [79] |
| Dexamethasone (1000-3000 ng/mL) | Dose-dependent upregulation of PGE2, IDO, and HLA-G | Enhanced stemness markers (Oct-4, Sox-2, Nanog); Improved immunosuppression in mixed lymphocyte reaction | [84] |
Experimental Design and Methodological Protocols
Standardized Preconditioning Workflow
The following diagram illustrates a generalized experimental workflow for preconditioning MSCs with hypoxia and inflammatory cytokines, compiled from multiple established protocols [79] [80] [83]:
Detailed Protocol for Combined Hypoxia and Inflammatory Preconditioning
Based on a 2023 study investigating umbilical cord-derived MSCs (UC-MSCs), the following protocol details the specific steps for effective combined preconditioning [79]:
Step 1: MSC Culture and Expansion
- Isplicate MSCs from tissue source (umbilical cord Wharton's jelly, bone marrow, or adipose tissue)
- Culture in complete medium (DMEM/F12 supplemented with appropriate serum replacement)
- Maintain at standard conditions (37°C, 5% CO₂, 95% air - normoxia)
- Use cells at passage 4 at 70-80% confluence for preconditioning experiments
Step 2: Cytokine Cocktail Preparation
- Prepare a mixture of pro-inflammatory cytokines including:
- Interferon-γ (IFN-γ): Concentration range 10-50 ng/mL
- Tumor necrosis factor-α (TNF-α): Concentration range 10-20 ng/mL
- Interleukin-1β (IL-1β): Concentration range 5-10 ng/mL
- Add cytokine mixture directly to culture medium
Step 3: Hypoxic Exposure
- Immediately transfer culture to three-gas incubator pre-set to:
- 2% O₂
- 5% CO₂
- 93% N₂
- 37°C
- Maintain cells under these conditions for 24 hours
Step 4: Post-Preconditioning Processing
- After 24 hours, harvest primed MSCs (designated PUC-MSCs in original study)
- Assess cell viability, typically exceeding 90% in validated protocols
- Use immediately for transplantation or further analysis
Quality Control and Characterization
Comprehensive assessment of preconditioned MSCs should include [79] [3]:
- Viability and proliferation: Trypan blue exclusion and MTT assays
- Surface marker profiling: Flow cytometry confirmation of CD73, CD90, CD105 expression (≥95%) and absence of hematopoietic markers (CD34, CD45, CD14/CD11b, CD79α/CD19, HLA-DR; ≤2%)
- Mitochondrial function: Assessment of membrane potential and integrity
- Immunomodulatory potential: Measurement of IDO activity, PGE2 production, and other soluble factors
- Functional assays: Coculture with peripheral blood mononuclear cells (PBMCs) or natural killer (NK) cells at optimal ratios (typically 3:1 PBMC:MSC)
Molecular Signaling Pathways in MSC Preconditioning
The enhanced therapeutic properties of preconditioned MSCs are mediated through specific molecular signaling pathways that are activated by hypoxia and inflammatory stimuli. The following diagram illustrates the key pathways involved and their interconnections:
The molecular mechanisms depicted above translate into specific functional enhancements that improve the therapeutic performance of preconditioned MSCs. Key pathway interactions include:
HIF-1α mediated adaptations: Under hypoxic conditions, HIF-1α stabilization leads to transcriptional activation of genes involved in glucose metabolism, angiogenesis, and cell survival, particularly VEGF [82]. This enhances MSC survival in low-oxygen environments and promotes trophic support for damaged tissues.
Cytokine-mediated immunomodulation: Inflammatory cytokines activate STAT and NF-κB signaling pathways, which in turn upregulate immunomodulatory molecules like IDO, TSG-6, and PGE2 [80] [83]. IDO activity is particularly crucial for T-cell suppression through tryptophan depletion and kynurenine production [80].
Integrated stress response: The combination of hypoxia and inflammatory signaling activates integrated cellular stress responses that enhance MSC resilience while simultaneously boosting paracrine functions [79]. This includes upregulation of antioxidant defenses through the Nrf2/ARE pathway [82].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for MSC Preconditioning Experiments
| Reagent/Category | Specific Examples | Function in Preconditioning | Application Notes |
|---|---|---|---|
| Pro-inflammatory Cytokines | IFN-γ, TNF-α, IL-1β, IL-17 | Activate immunomodulatory pathways; Enhance immunosuppressive potential | Optimal concentrations: 10-50 ng/mL; Cocktail approaches often most effective [79] [83] |
| Hypoxia Mimetics | Cobalt chloride (CoCl₂), Deferoxamine (DFO) | Stabilize HIF-1α; Simulate hypoxic conditions | Alternative to hypoxic chambers; Concentration-dependent effects [82] |
| Culture Media & Supplements | DMEM/F12, MesenCult, FBS, Serum replacements | Maintain cell viability; Support priming process | Serum-free alternatives reduce variability [79] |
| Analysis Kits | IDO activity assays, PGE2 ELISA, VEGF ELISA | Quantify immunomodulatory molecule production | Essential for validating priming efficacy [79] [84] |
| Flow Cytometry Antibodies | CD73, CD90, CD105, CD34, CD45, HLA-DR | Verify MSC phenotype post-priming | Confirmation of marker expression critical for quality control [79] [3] |
| Cell Culture Equipment | Three-gas incubators, Hypoxia chambers | Maintain precise O₂, CO₂, N₂ levels | Required for physiological hypoxia models [79] |
Clinical Translation and Current Status
The promising preclinical data on MSC preconditioning has begun to translate into early-stage clinical trials. As of 2024, clinical evidence supporting preconditioning strategies remains limited to a few Phase I trials, most of which are still in progress [81]. The current clinical landscape includes:
Completed and Ongoing Trials:
- Safety studies of IFN-γ-primed MSCs have established initial safety profiles for cytokine-preconditioned approaches [81]
- Glycoengineered MSCs in advanced osteoporosis demonstrated decreased pain scores and no new fractures in a small clinical trial [81]
- Combined preconditioning approaches are now entering early-phase clinical testing based on robust preclinical efficacy data [79] [81]
Clinical Considerations:
- Safety profile: Preconditioned MSCs maintain the favorable safety characteristics of naive MSCs, with no significant additional safety concerns identified to date [81]
- Potency enhancement: Preconditioning may help mitigate the heterogeneity of MSC lots from different donors, improving consistency in therapeutic outcomes [81]
- Application-specific optimization: The optimal preconditioning strategy depends on the specific clinical application, requiring elucidation of the primary mechanism of action for each therapeutic context [81]
Preconditioning of MSCs with hypoxia and inflammatory cytokines represents a powerful strategy to enhance their therapeutic efficacy by mimicking the challenging in vivo environment they encounter after transplantation. The molecular mechanisms involving HIF-1α stabilization, STAT/NF-κB signaling, and subsequent upregulation of immunomodulatory factors have been well-characterized in preclinical models [79] [80] [82]. Standardized protocols now enable researchers to consistently generate primed MSCs with enhanced survival, immunomodulatory capacity, and tissue-reparative functions.
Future directions in this field include optimizing combination approaches, developing more precise cytokine cocktails for specific disease applications, and addressing manufacturing challenges for clinical-scale production [81]. As preconditioning strategies continue to evolve, they hold significant promise for unlocking the full therapeutic potential of MSC-based therapies across a wide range of clinical applications, particularly in immune-mediated inflammatory diseases and degenerative conditions [79] [2] [81].
The therapeutic potential of mesenchymal stem cells (MSCs) in regenerative medicine is substantially limited by critical challenges post-transplantation, including poor cell survival, inefficient homing to injury sites, and variable secretory functions. This technical review comprehensively examines advanced genetic engineering strategies designed to overcome these limitations. We provide a detailed analysis of molecular targets and methodologies for enhancing MSC efficacy, with a specific focus on comparative differences between bone marrow-derived MSCs (BM-MSCs) and adipose-derived MSCs (AD-MSCs). The review incorporates structured experimental protocols, key reagent specifications, and visual workflow diagrams to serve as a comprehensive resource for researchers and drug development professionals working in advanced therapeutic medicinal products.
Mesenchymal stem cells (MSCs) represent a promising platform for regenerative medicine due to their multipotent differentiation capacity, immunomodulatory properties, and paracrine secretion capabilities [2]. According to the International Society for Cellular Therapy (ISCT), MSCs are defined by their plastic adherence, specific surface marker expression (CD73, CD90, CD105 ≥95%; CD34, CD45, CD14, CD19, HLA-DR ≤2%), and tri-lineage differentiation potential [2] [20]. Despite their therapeutic potential, clinical applications face significant hurdles, with studies demonstrating that less than 5% of intravenously administered MSCs survive in target tissues beyond four weeks post-transplantation [85]. This attrition represents a major bottleneck in realizing the full therapeutic potential of MSC-based therapies.
The homing process—whereby MSCs navigate to sites of injury or inflammation—is a multi-step cascade involving initial tethering by selectins, activation by cytokines, arrest by integrins, diapedesis via matrix remodelers, and extravascular migration toward chemokine gradients [86]. Each stage presents opportunities for therapeutic enhancement through genetic modification. Furthermore, significant functional differences exist between MSC sources: AD-MSCs demonstrate superior proliferation rates and adipogenic differentiation, while BM-MSCs exhibit enhanced osteogenic and chondrogenic capacity [20]. Understanding these source-specific characteristics is crucial for designing targeted genetic engineering approaches.
While MSCs from various sources share fundamental characteristics, their functional properties, differentiation potential, and secretory profiles vary significantly based on their tissue of origin. These differences have profound implications for their therapeutic application and for selecting the most appropriate source for specific clinical indications.
Table 1: Comparative Characteristics of BM-MSCs and AD-MSCs
| Characteristic | BM-MSCs | AD-MSCs | Clinical Implications |
|---|---|---|---|
| Collection Procedure | Invasive bone marrow aspiration [20] | Minimally invasive lipoaspiration [23] [20] | AD-MSCs offer easier harvest and greater tissue availability |
| Cell Yield | 0.001–0.01% of harvested marrow [20] | Significantly higher yield [23] | AD-MSCs require less expansion time |
| Proliferation Rate | Slower proliferation, signs of early senescence [20] | Faster proliferation [23] [20] | AD-MSCs更适合大规模扩增 |
| Osteogenic Potential | Superior osteogenic differentiation [20] | Reduced osteogenic capacity [20] | BM-MSCs preferred for bone regeneration |
| Chondrogenic Potential | Enhanced chondrogenesis [20] | Reduced chondrogenic capacity [20] | BM-MSCs preferred for cartilage repair |
| Adipogenic Potential | Reduced adipogenesis [20] | Superior adipogenic differentiation [20] | AD-MSCs ideal for adipose tissue engineering |
| Immunomodulatory Effects on B-cells | Moderate suppression of immunoglobulin production [87] | Strong inhibition of B-cell immunoglobulin production [87] | AD-MSCs potentially superior for B-cell mediated disorders |
| Surface Marker Variations | High expression of Stro-1 [20] | Low expression of Stro-1, high expression of CD49d [20] | Different marker profiles may affect homing capabilities |
| Stress Resilience | Moderate resilience to oxidative stress/hypoxia [23] | Enhanced survival under oxidative stress/hypoxia [23] | AD-MSCs may better withstand inflammatory environments |
Secretory Profile Variations
The paracrine functions of MSCs—mediated through growth factors, cytokines, and extracellular vesicles—are increasingly recognized as their primary therapeutic mechanism [88]. Both BM-MSCs and AD-MSCs secrete a diverse array of bioactive molecules, but with notable differences in their secretory profiles. AD-MSCs demonstrate enhanced secretion of angiogenic factors like VEGF and immunomodulatory cytokines such as IL-10, which promotes M2 macrophage polarization and T regulatory cell proliferation [23]. Conversely, BM-MSCs exhibit stronger secretion of osteogenic and chondrogenic factors. These differential secretory profiles provide a rationale for selecting specific MSC sources for particular disease contexts and for genetic engineering approaches to enhance desirable secretions.
Molecular Mechanisms of MSC Homing
The Multi-Step Homing Cascade
The homing of MSCs to sites of injury is a complex, multi-step process that mirrors leukocyte trafficking but with distinct molecular mechanisms. Understanding this cascade is essential for developing effective genetic engineering strategies to enhance homing efficiency.
The homing cascade initiates with tethering and rolling mediated by selectins (P-selectin and E-selectin) on endothelial cells interacting with corresponding ligands on MSCs [86]. This is followed by activation through chemokines such as stromal-derived factor-1 (SDF-1) binding to G-protein coupled receptors on MSCs, triggering intracellular signaling that activates integrins. During arrest and adhesion, activated integrins including VLA-4 (Very Late Antigen-4) mediate firm adhesion to endothelial adhesion molecules like VCAM-1 (Vascular Cell Adhesion Molecule-1) and ICAM-1 (Intercellular Adhesion Molecule-1) [86]. Diapedesis then occurs via paracellular or transcellular migration facilitated by matrix metalloproteinases (MMPs) that degrade the extracellular matrix. Finally, extravascular migration directs MSCs toward chemokine gradients (SDF-1, HGF) within injured tissues [86].
Key Molecular Targets for Genetic Enhancement
Several molecular pathways present promising targets for genetic engineering to enhance homing efficiency:
- CXCR4/SDF-1 Axis: The chemokine receptor CXCR4 and its ligand SDF-1 represent the most potent chemotactic axis for MSC homing. Genetic modification to overexpress CXCR4 enhances MSC migration toward SDF-1 gradients typically elevated in injured tissues [86].
- Integrins: Overexpression of integrin subunits (especially α4 integrin component of VLA-4) strengthens MSC adhesion to endothelial VCAM-1, facilitating firm arrest [86].
- Matrix Remodeling Enzymes: Engineering MSCs to express optimized levels of MMPs (particularly MMP-2 and MMP-9) enhances their transendothelial migration capacity without promoting excessive matrix degradation [86].
Genetic Engineering Strategies to Enhance MSC Functions
Approaches for Improving Survival and Engraftment
Enhancing MSC survival post-transplantation is critical for therapeutic efficacy. Genetic strategies focus on combating apoptosis and cellular stress encountered in the hostile in vivo environment.
Table 2: Genetic Engineering Strategies for Enhanced MSC Survival
| Target Pathway | Genetic Approach | Mechanism of Action | Outcome | Evidence Level |
|---|---|---|---|---|
| Akt/PKB Signaling | Overexpress constitutively active Akt | Inhibits mitochondrial apoptosis pathway; enhances nutrient utilization | ≥2-fold increase in survival under hypoxic conditions [85] | Preclinical (in vivo) |
| BCL-2 Family | Overexpress anti-apoptotic BCL-2 or BCL-xL | Blocks cytochrome c release; prevents caspase activation | Reduces apoptosis by 40-60% in inflammatory environments [85] | Preclinical (in vitro) |
| HIF-1α Stabilization | Express oxygen-stable HIF-1α variant | Activates glycolytic metabolism; upregulates pro-survival genes | Enhances survival in ischemic tissues by 30-50% [85] | Preclinical (in vivo) |
| SOD2 Overexpression | Enhance mitochondrial superoxide dismutase | Reduces reactive oxygen species; prevents oxidative damage | Improves resistance to oxidative stress by 70% [85] | Preclinical (in vitro) |
| Telomerase Reverse Transcriptase (TERT) | Inducible TERT expression | Extends telomeres; delays replicative senescence | Prolongs proliferative capacity by 40% without tumorigenesis [2] | Preclinical (in vitro) |
Experimental Protocol: Genetic Modification with Akt for Enhanced Survival
Objective: To enhance MSC survival through lentiviral-mediated overexpression of constitutively active Akt.
Materials:
- Primary human BM-MSCs or AD-MSCs (passage 3-5)
- Lentiviral vector encoding constitutively active Akt (myr-Akt)
- Control lentiviral vector (empty or GFP-only)
- Polybrene (8μg/mL)
- Puromycin or appropriate selection antibiotic
- MSC expansion medium: DMEM/F12 with 10% FBS, 1% penicillin/streptomycin
- Apoptosis induction medium: serum-free DMEM with 200μM H₂O₂
- Annexin V-FITC/PI apoptosis detection kit
- Western blot equipment for Akt and phospho-Akt detection
Methodology:
- Cell Preparation: Culture MSCs to 70% confluence in T75 flasks. Harvest using standard trypsin/EDTA protocol and seed in 6-well plates at 1×10⁵ cells/well.
- Viral Transduction: Incubate MSCs with lentiviral vectors (multiplicity of infection 10-50) in presence of polybrene for 24 hours.
- Selection: Replace medium with fresh expansion medium containing puromycin (1-2μg/mL) for 7 days to select successfully transduced cells.
- Validation: Confirm Akt overexpression via Western blot analysis using anti-Akt and anti-phospho-Akt antibodies.
- Survival Assay: Subject modified and control MSCs to serum-free conditions or H₂O₂ treatment for 48 hours. Quantify apoptosis using Annexin V-FITC/PI staining followed by flow cytometry analysis.
- Functional Assessment: Compare metabolic activity (MTT assay), caspase activation (caspase-3/7 assay), and mitochondrial membrane potential (JC-1 staining) between modified and control cells.
Expected Outcomes: Akt-modified MSCs should demonstrate significantly reduced apoptosis (≥40% reduction in Annexin V-positive cells) and enhanced metabolic activity under stress conditions compared to control MSCs.
Homing Enhancement Through CXCR4 Modulation
The CXCR4/SDF-1 axis represents the most promising target for enhancing MSC homing. Genetic engineering approaches have focused on both increasing CXCR4 expression and modifying its regulatory mechanisms.
Experimental Protocol: CXCR4 Overexpression for Enhanced Homing
Objective: To enhance MSC homing capacity through lentiviral-mediated CXCR4 overexpression.
Materials:
- Primary human AD-MSCs (passage 3-4)
- Lentiviral vector encoding human CXCR4
- Control lentiviral vector (GFP-only)
- Recombinant human SDF-1α
- Transwell migration chambers (5μm pore size)
- Flow cytometry equipment with anti-CXCR4 antibody
- In vivo imaging system (IVIS) for cell tracking
Methodology:
- Cell Preparation and Transduction: Culture AD-MSCs to 70% confluence. Transduce with CXCR4 or control lentiviral vectors as described in Section 4.2.
- Validation of CXCR4 Expression: Confirm CXCR4 overexpression using flow cytometry (anti-CXCR4 antibody) 72 hours post-transduction.
- In Vitro Migration Assay:
- Place serum-free medium in lower chamber with or without SDF-1α (100ng/mL).
- Seed 2.5×10⁴ transduced MSCs in serum-free medium in upper chamber.
- Incubate for 6 hours at 37°C.
- Count migrated cells on lower membrane surface after crystal violet staining.
- Calculate migration index: (cells migrated toward SDF-1 / cells migrated toward medium alone).
- In Vivo Homing Assessment:
- Label transduced MSCs with near-infrared dye (DIR).
- Inject 1×10⁶ cells intravenously into mouse model of myocardial infarction.
- Monitor homing to infarcted heart using IVIS at 24, 48, and 72 hours post-injection.
- Quantify fluorescence intensity in heart tissue relative to control organs.
Expected Outcomes: CXCR4-overexpressing MSCs should demonstrate ≥2-fold increased migration toward SDF-1 in vitro and ≥3-fold enhanced homing to infarcted myocardium compared to control MSCs.
Enhancing Secretory Functions Through Genetic Modification
Strategic Enhancement of Paracrine Factor Production
The therapeutic benefits of MSCs are largely mediated through their paracrine secretions, including growth factors, cytokines, and extracellular vesicles [88]. Genetic engineering can be employed to enhance the production of specific therapeutic factors.
Table 3: Genetic Modification Strategies for Enhanced Secretory Functions
| Target Molecule | Therapeutic Rationale | Genetic Approach | Application Context | Outcome |
|---|---|---|---|---|
| VEGF | Promotes angiogenesis; enhances tissue revascularization | Lentiviral overexpression | Ischemic diseases (myocardial infarction, critical limb ischemia) | ≥2-fold increase in capillary density in ischemic tissues [23] |
| IL-10 | Potent anti-inflammatory; promotes M2 macrophage polarization | Plasmid transfection or viral vector | Inflammatory disorders (graft-versus-host disease, rheumatoid arthritis) | 60-70% reduction in pro-inflammatory cytokines (TNF-α, IL-6) [23] |
| HGF | Mitogenic for hepatocytes; anti-fibrotic; enhances regeneration | Lentiviral or AAV vector | Liver diseases (cirrhosis, acute liver failure) | Significant reduction in fibrosis and improved liver function [85] |
| BDNF | Neurotrophic factor; supports neuronal survival and differentiation | Lentiviral overexpression | Neurological disorders (stroke, spinal cord injury) | Enhanced neuronal survival and functional recovery [2] |
| miR-125a-3p | Inhibits proliferation and activation of Th2 cells (via exosomes) | miRNA overexpression | Allergic and atopic conditions (atopic dermatitis) | Reduced pathological symptoms in atopic dermatitis models [23] |
| miR-21-3p | Promotes keratinocyte and endothelial cell proliferation | Engineered exosomes | Wound healing applications | Accelerated wound closure in diabetic models [23] |
Experimental Protocol: Engineering MSCs for Enhanced VEGF Secretion
Objective: To genetically modify MSCs for sustained VEGF secretion to promote angiogenesis.
Materials:
- Primary human BM-MSCs (passage 3-4)
- Lentiviral vector encoding human VEGF₁₆₅
- Control lentiviral vector (GFP-only)
- VEGF ELISA kit
- Human umbilical vein endothelial cells (HUVECs)
- Matrigel matrix
- Tube formation assay reagents
- Mouse hindlimb ischemia model
Methodology:
- Genetic Modification: Transduce BM-MSCs with VEGF or control lentiviral vectors as described in Section 4.2.
- VEGF Secretion Quantification: Culture transduced MSCs for 48 hours, collect conditioned medium, and quantify VEGF concentration using ELISA.
- In Vitro Angiogenic Assay:
- Concentrate conditioned medium from modified and control MSCs.
- Seed HUVECs on Matrigel-coated plates with conditioned medium.
- Incubate for 6-8 hours and quantify tube formation (number of branches, tube length).
- In Vivo Therapeutic Assessment:
- Induce hindlimb ischemia in immunodeficient mice via femoral artery ligation.
- Intramuscularly inject 5×10⁵ VEGF-modified or control MSCs into ischemic muscle.
- Assess perfusion recovery using laser Doppler imaging at days 0, 7, 14, and 28.
- Harvest tissue at day 28 for capillary density quantification (CD31 immunohistochemistry).
Expected Outcomes: VEGF-modified MSCs should secrete ≥5-fold higher VEGF levels, induce enhanced HUVEC tube formation in vitro (≥2-fold increase in branches), and significantly improve perfusion recovery and capillary density in ischemic hindlimbs compared to control MSCs.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for MSC Genetic Engineering
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Viral Vectors | Lentivirus, Adenovirus (AdV), Adeno-associated virus (AAV) | Stable (lentivirus) or transient (AdV, AAV) genetic modification | Lentivirus for long-term expression; AAV for reduced immunogenicity |
| Non-Viral Transfection | Electroporation, Lipofection, Nanoparticles | Plasmid DNA delivery without viral components | Lower efficiency but reduced safety concerns |
| Selection Antibiotics | Puromycin, G418, Hygromycin | Selection of successfully transduced cells | Concentration must be optimized for each MSC type |
| Cell Tracking Agents | GFP/luciferase vectors, DIR dye, Quantum dots | In vitro and in vivo cell tracking | Consider trade-offs between sensitivity and resolution |
| Migration Assay Systems | Transwell chambers, μ-Slide Chemotaxis | Quantitative assessment of migratory capacity | Pore size (5-8μm) optimized for MSC migration |
| Characterization Antibodies | CD73, CD90, CD105, CD34, CD45, HLA-DR | MSC phenotype validation post-modification | Essential for confirming MSC identity after genetic manipulation |
| Cytokines/Chemokines | Recombinant SDF-1, VEGF, HGF | In vitro functional assays for homing/secretory capacity | Quality and bioactivity critical for reliable results |
| Animal Disease Models | Myocardial infarction, Hindlimb ischemia, Skin wounds | In vivo assessment of therapeutic efficacy | Model selection should match clinical application |
Genetic engineering strategies offer powerful approaches to overcome the fundamental limitations of MSC-based therapies, including poor survival, inefficient homing, and variable secretory functions. The selection of appropriate MSC sources—whether BM-MSCs or AD-MSCs—should be guided by their inherent biological differences and the specific therapeutic application. The genetic modification approaches detailed in this review, targeting key pathways in survival (Akt, BCL-2), homing (CXCR4, integrins), and secretory functions (VEGF, IL-10, microRNAs), provide researchers with validated methodologies to enhance MSC therapeutic potential. As the field advances, the integration of more sophisticated gene regulation systems, including inducible promoters and miRNA-based fine-tuning, will further refine our ability to optimize MSC function for specific clinical applications. The ongoing challenge remains balancing enhanced efficacy with strict safety profiles, particularly as these engineered cell products move toward clinical translation.
Mesenchymal stem cell (MSC) therapy has undergone a paradigm shift with the emergence of extracellular vesicles (EVs) and exosomes as powerful cell-free therapeutic agents. While MSCs themselves have demonstrated significant potential in regenerative medicine due to their multipotent differentiation capacity, immunomodulatory properties, and paracrine activities, their therapeutic application faces challenges including poor engraftment, potential immunogenicity, and risks of tumorigenicity or thrombosis [89] [2]. Research has progressively revealed that many of the therapeutic benefits of MSCs are mediated through their secretome, particularly through EVs and exosomes, which carry bioactive molecules from parent cells to recipient cells [18] [78]. These nanoparticles offer comparable efficacy with lower immunogenicity, greater stability, and reduced safety concerns, positioning them as next-generation therapeutics [89] [78].
This technical overview examines the current landscape of MSC-derived EVs and exosomes, with a specific focus on their characterization, therapeutic mechanisms, clinical translation, and standardized methodologies. The content is framed within the broader context of MSC sources, including bone marrow, adipose tissue, umbilical cord, and other tissues, highlighting how source selection influences therapeutic potential and clinical applications.
Biological Foundations of MSC-Derived EVs and Exosomes
Defining Extracellular Vesicles and Exosomes
Extracellular vesicles are membrane-bound nanoparticles released by virtually all cell types, functioning as crucial mediators of intercellular communication. The EV landscape encompasses several subtypes classified by biogenesis and size: apoptotic vesicles (1,000–5,000 nm), microvesicles (100–1,000 nm), and exosomes (30–200 nm) [18]. Exosomes, a specific EV subtype, form intracellularly via the endosomal pathway where cellular cargo is internalized into early endosomes that mature into multivesicular bodies (MVBs); subsequent fusion of MVBs with the plasma membrane releases exosomes into the extracellular environment [78].
According to the International Society for Extracellular Vesicles (ISEV), the term "EVs" encompasses all membrane-bound particles released by cells, including both microvesicles and exosomes, reflecting the practical challenges in distinguishing these populations [90]. MSC-derived EVs and exosomes transport a diverse molecular cargo including proteins, lipids, mRNAs, miRNAs, and DNA that reflect their cellular origin and mediate their biological functions [90] [91].
MSCs can be isolated from various adult and perinatal tissues, each with distinct advantages and limitations:
- Bone Marrow-MSCs (BM-MSCs): The most extensively studied source, known for high differentiation potential and strong immunomodulatory effects, but requiring invasive harvesting with limited cell yield (approximately 0.01-0.001% of bone marrow cells) [2] [3].
- Adipose Tissue-MSCs (AD-MSCs): Readily available from liposuction procedures with higher yields (up to 1 billion cells from 300g of tissue), offering faster proliferation and comparable therapeutic properties to BM-MSCs [18] [3].
- Umbilical Cord-MSCs (UC-MSCs): Isolated from Wharton's jelly, these cells exhibit enhanced proliferation, lower immunogenicity, and are suitable for allogeneic transplantation [2] [3].
- Other Sources: Placenta, menstrual blood, dental pulp, and umbilical cord blood also provide MSCs with unique properties and fewer ethical concerns [3].
The biological functions and characteristics of MSC-EVs vary significantly in size, composition, and function depending on their tissue source, influencing their therapeutic efficacy for different diseases [89] [92].
Key Signaling Pathways and Therapeutic Mechanisms
MSC-derived exosomes modulate recipient cell behavior through several critical signaling pathways. The following diagram illustrates the primary mechanisms through which MSC-exosomes exert their therapeutic effects on target cells:
Therapeutically, MSC-EVs have demonstrated broad applications across multiple organ systems by promoting tissue repair, modulating immune responses, and reducing inflammation through several key mechanisms:
Immunomodulation: MSC-EVs inhibit proinflammatory macrophage polarization while promoting anti-inflammatory macrophages, regulate T helper cell proliferation and differentiation, and suppress B cell development [90] [23]. Specific miRNAs in AD-MSC exosomes (e.g., miR-125a-3p) inhibit proliferation and activation of Th2 cells, relevant for inflammatory skin diseases [23].
Tissue Regeneration: EVs enhance angiogenesis through vascular endothelial growth factor (VEGF) and other factors, promote epithelial-mesenchymal transition, and exert antiapoptotic effects [90]. They activate pro-regenerative pathways including Wnt/β-catenin and PI3K/Akt signaling, stimulating resident stem and progenitor cells [18] [78].
Anti-fibrotic Effects: In disease models such as liver fibrosis, MSC-exosomes deliver regulatory miRNAs that reduce collagen production and inhibit fibrotic pathway activation [91].
Clinical Translation and Trial Data
Analysis of Registered Clinical Trials
A comprehensive review of clinical trials registered between 2014 and 2024 provides insights into the current landscape of MSC-EV therapeutics. Data collected from ClinicalTrials.gov, the Chinese Clinical Trial Registry, and the Cochrane Register of Studies identified 66 eligible trials after screening [89]. The visual workflow below outlines the clinical trial collection and analysis process:
Administration Routes and Dose Optimization
Clinical trials have identified two predominant administration methods: intravenous infusion and aerosolized inhalation, with the latter being particularly prominent for respiratory diseases including COVID-19 [89]. Notably, dose-effect relationships reveal that nebulization therapy achieves therapeutic effects at approximately 10⁸ particles, significantly lower than doses required for intravenous administration, suggesting a route-dependent effective dose window [89].
The table below summarizes key quantitative findings from clinical trials and research on MSC-EV dosing and administration:
Table 1: Clinical Administration Routes and Dose Ranges for MSC-EVs and Exosomes
| Administration Route | Therapeutic Dose Range | Primary Clinical Applications | Advantages | Limitations |
|---|---|---|---|---|
| Intravenous Infusion | Higher doses required (typically >10⁹ particles) | Systemic diseases, acute inflammation, neurological disorders | Systemic distribution, reaches multiple organs | Potential rapid clearance, dose-dependent side effects |
| Aerosolized Inhalation | ~10⁸ particles | Respiratory diseases (COVID-19, ARDS, pulmonary fibrosis) | Targeted lung delivery, lower effective dose | Primarily limited to respiratory applications |
| Local Injection | Varies by tissue target | Orthopedic injuries, dermatological applications, ovarian disorders | Direct tissue targeting, minimal systemic exposure | Invasive, limited to accessible tissues |
| Topical Application | Formulation-dependent | Wound healing, dermatological conditions, corneal repair | Non-invasive, patient self-administration possible | Formulation stability challenges |
Disease Targets and Clinical Applications
MSC-EVs and exosomes have been investigated across a broad spectrum of medical conditions. The table below summarizes key disease targets and observed therapeutic effects:
Table 2: Clinical Applications of MSC-Derived EVs and Exosomes by Disease Area
| Disease Category | Specific Conditions | MSC Sources | Reported Therapeutic Effects |
|---|---|---|---|
| Respiratory Diseases | COVID-19, ARDS, pulmonary fibrosis | BM, AD, UC | Reduced inflammation, improved lung function, alveolar repair |
| Neurological Disorders | Stroke, Alzheimer's disease, spinal cord injury | BM, UC | Neuroprotection, reduced infarct volume, functional recovery |
| Autoimmune Conditions | Graft-versus-host disease, psoriasis, atopic dermatitis | AD, BM, UC | Immunomodulation, reduced clinical scores, serum biomarkers |
| Liver Diseases | Liver fibrosis, acute liver injury | BM, AD, UC | Anti-fibrotic effects, hepatocyte regeneration, inflammation reduction |
| Musculoskeletal Disorders | Osteoarthritis, bone/cartilage defects | BM, AD | Cartilage protection, pain reduction, tissue regeneration |
| Gynecological Conditions | Premature ovarian insufficiency, intrauterine adhesions | UC, menstrual blood, endometrium | Endometrial repair, ovarian function restoration |
Experimental Protocols and Methodologies
MSC-EV Isolation and Purification
Standardized protocols for isolating and purifying EVs and exosomes remain a significant challenge in the field. The following workflow outlines key methodological approaches:
The most common isolation methods include:
- Differential Ultracentrifugation: The historical gold standard involving sequential centrifugation steps to separate EVs based on size and density [89] [92].
- Size-Based Techniques: Size exclusion chromatography and tangential flow filtration offer improved scalability and preservation of EV integrity [78].
- Polymer-Based Precipitation: Utilizes hydrophilic polymers to precipitate EVs, though may co-precipitate contaminants [92].
Characterization and Quality Control
Comprehensive characterization of MSC-EVs is essential for quality control and batch consistency. The International Society for Cellular Therapy (ISCT) and ISEV have established minimum standards for MSC and EV characterization respectively [89] [3]. Key characterization assays include:
- Nanoparticle Tracking Analysis (NTA): Determines particle size distribution and concentration [89].
- Transmission Electron Microscopy (TEM): Visualizes EV morphology and ultrastructure [89].
- Flow Cytometry: Detects surface protein markers (CD9, CD63, CD81 for exosomes; CD73, CD90, CD105 for MSC origin) [89] [3].
- Western Blotting: Confirms presence of EV-enriched proteins and absence of contaminants.
- Proteomic and Genomic Analysis: Mass spectrometry and RNA sequencing characterize molecular cargo [90].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for MSC-EV Studies
| Reagent Category | Specific Examples | Research Function | Technical Considerations |
|---|---|---|---|
| Cell Culture Media | Serum-free MSC media, Xeno-free supplements | MSC expansion and EV production | Eliminates bovine EV contamination; maintains MSC phenotype |
| EV Isolation Kits | Polymer-based precipitation kits, Size exclusion columns | Rapid EV isolation from conditioned media | Varying purity and recovery rates; potential co-isolation of contaminants |
| Characterization Antibodies | Anti-CD9, CD63, CD81, CD73, CD90, CD105 | EV validation and source confirmation | Requires multiplex approach; species-specific validation needed |
| Visualization Reagents | Lipophilic dyes (PKH67, DiI), Membrane labels | EV tracking and uptake studies | Potential dye aggregation; appropriate controls essential |
| RNA/Protein Analysis Kits | miRNA extraction kits, Multiplex immunoassays | Cargo analysis and functional characterization | Small RNA focus; sensitivity thresholds vary |
| Engineering Tools | Transfection reagents, CRISPR/Cas9 systems | EV modification for enhanced targeting | Optimization required for different MSC sources |
Engineering Strategies for Enhanced Therapeutics
Engineering MSC-derived exosomes enhances their therapeutic potential through improved targeting, cargo loading, and pharmacokinetics. The primary engineering approaches include:
Genetic Modification of Parent MSCs: Engineering MSCs to overexpress therapeutic miRNAs, cytokines, or targeting ligands, which are then incorporated into secreted exosomes [91]. For liver disease treatment, MSC-exosomes engineered to carry specific miRNAs (e.g., miR-122, miR-181) demonstrated enhanced anti-fibrotic and regenerative effects in preclinical models [91].
Direct Exosome Modification: Post-isolation engineering through electroporation, sonication, or extrusion to load therapeutic cargo, or surface modification with targeting peptides or antibodies to enhance tissue-specific delivery [90] [91].
Preconditioning Strategies: Exposure of MSCs to specific microenvironmental conditions (hypoxia, inflammatory cytokines, or pharmacological agents) to modify EV cargo and enhance therapeutic efficacy [92].
Current Challenges and Future Directions
Despite significant progress, several challenges impede the clinical translation of MSC-EV therapeutics:
Manufacturing and Standardization: Scalable production of GMP-compliant EVs with consistent potency remains technically challenging [90] [78]. Transition from flask-based culture to closed bioreactor systems is essential for clinical-grade production [78].
Analytical and Potency Assays: Lack of standardized potency assays, biodistribution studies, and dosing metrics complicates regulatory evaluation [89] [78]. Rapid in vivo clearance and donor variability present additional hurdles [78].
Regulatory Frameworks: No drug regulatory authorities have issued specific technical evaluation guidelines for EV-based drugs, creating uncertainty in development pathways [90].
Future directions include advancing engineering approaches for enhanced targeting and payload delivery, developing integrated manufacturing platforms, establishing universally accepted potency metrics, and conducting larger controlled clinical trials to definitively establish efficacy across disease indications [90] [78] [91].
MSC-derived extracellular vesicles and exosomes represent a transformative approach in regenerative medicine, combining the therapeutic benefits of cellular therapies with the advantages of acellular systems. As the field progresses from preclinical validation to clinical application, addressing challenges in manufacturing standardization, analytical characterization, and regulatory alignment will be crucial for realizing the full potential of these promising therapeutics. Continued research into MSC source selection, EV engineering, and mechanism of action will further advance the field toward effective clinical applications across a broad spectrum of diseases.
The field of Mesenchymal Stem Cell (MSC) research stands at a pivotal crossroads. With over 600 clinical trials registered and a market value projected to grow from USD 3.87 billion in 2025 to USD 13.49 billion by 2035, the therapeutic potential of MSCs is undeniable [93] [69]. These cells, derived from diverse sources including bone marrow, adipose tissue, and umbilical cord, have demonstrated promising results in treating conditions ranging from autoimmune diseases and orthopedic injuries to inflammatory disorders [2]. However, this promise remains largely unfulfilled at a broader clinical level. A fundamental challenge persists: the inability to consistently manufacture homogeneous MSC populations with known efficacy for specific disease indications that yield predictable and reproducible patient outcomes [93].
This challenge stems primarily from two interconnected problems: the inherent heterogeneity of MSC populations and the lack of standardized, predictive potency assays. MSCs are highly heterogeneous in terms of their biological characteristics, influenced by factors including donor age and health, tissue source, isolation methods, and culture conditions [94] [95]. This heterogeneity presents extraordinary challenges to the approvability of MSC-based therapies, as comparability and consistency between manufacturing batches become difficult to ensure [94]. While the International Society for Cellular Therapy (ISCT) has established minimal criteria for defining MSCs (plastic adherence, specific surface marker expression, and tri-lineage differentiation potential), these criteria are insufficient for predicting therapeutic efficacy [2] [96]. The lack of metrics that discriminate functional differences between MSC populations further confounds efforts to select the most suitable populations for a given disease indication [93]. This whitepaper examines the current state of MSC standardization and potency assays, provides detailed experimental methodologies, and proposes pathways toward resolving these critical unmet needs.
The therapeutic potential of MSCs is significantly influenced by their tissue of origin, with each source presenting unique advantages and limitations. Understanding this source-specific heterogeneity is fundamental to developing effective potency assays.
Table 1: Characteristics of MSC Sources Relevant to Therapeutic Applications
| Source | Key Advantages | Limitations | Primary Research Applications |
|---|---|---|---|
| Bone Marrow (BM-MSCs) | Most extensively studied; high differentiation potential; strong immunomodulatory effects [2] | Invasive harvesting procedure; decline in cell number and differentiation potential with donor age [2] [95] | Orthopedic injuries, graft-versus-host disease, hematopoietic support [2] |
| Adipose Tissue (AD-MSCs) | Easier to harvest with higher yields; comparable therapeutic properties to BM-MSCs [2] [97] | Variable quality based on donor metabolic health; potentially lower osteogenic potential compared to BM-MSCs [97] | Soft tissue regeneration, bone repair, cosmetic and reconstructive procedures [97] |
| Umbilical Cord (UC-MSCs) | Enhanced proliferation capacity; lower immunogenicity; suitable for allogeneic transplantation [2] | Ethical considerations; limited self-renewal compared to perinatal sources [2] [9] | Immunomodulatory applications, allogeneic banking, regenerative medicine [2] [9] |
| Dental Pulp (DP-MSCs) | Accessibility from medical waste; strong neurogenic and angiogenic potential [2] | Limited tissue volume; specialized isolation requirements [2] | Dental pulp regeneration, neurological applications [2] |
Factors Contributing to MSC Heterogeneity
Beyond tissue source, multiple variables contribute to MSC heterogeneity, creating substantial challenges for standardization:
- Donor-related factors: Age, health status, genetic background, and lifestyle factors significantly impact MSC functionality and potency [96]. MSCs from older donors often exhibit reduced proliferative capacity and differentiation potential compared to those from younger donors [95].
- Isolation and culture techniques: Methods such as enzymatic digestion, density gradient centrifugation, plastic adherence, and selection for specific surface markers (e.g., STRO-1, CD146, CD271) yield populations with different functional characteristics [9] [96]. Culture conditions, including media composition, oxygen tension, and passage number, further influence MSC properties [95].
- Functional heterogeneity: Even within clonal populations, MSCs exhibit significant functional diversity. Cells at the periphery of colonies express higher levels of proliferation-related genes (MKI67, PODXL), while interior MSCs show increased expression of extracellular matrix genes (VCAM1) [96].
This inherent variability necessitates the development of robust potency assays that can predict therapeutic efficacy despite biological differences between MSC batches.
Current Landscape of MSC Potency Assays
Potency assays are critical for ensuring that MSC-based therapies consistently produce their intended biological effects. Currently, three main approaches dominate the field, each with distinct advantages and limitations.
Univariate and Multivariate Assay Approaches
Table 2: Current Approaches to MSC Potency Assessment
| Assay Type | Measured Parameter | Advantages | Limitations |
|---|---|---|---|
| Univariate Angiogenic Assay | Endothelial tube formation induced by MSC-conditioned media; correlation with CXCL5, IL-8, VEGF levels [93] | Relatively simple; quantifiable; potential for surrogate marker testing | Focuses on single mechanism; may not predict efficacy in complex disease environments |
| Immunomodulatory Potency Assay | Suppression of T-cell proliferation at various effector-to-target ratios; calculation of mean suppression value [93] | Functional assessment of key MSC mechanism; broad dynamic range (27%–88%) | Does not correlate with cell viability or HLA-DR expression; may not reflect other immunomodulatory mechanisms |
| Inflammation-Specific Marker Assay | TSG-6 expression levels correlated with efficacy in reducing myeloperoxidase activity in corneal injury models [93] | Disease-specific relevance; clear biomarker correlation | Limited to acute inflammatory diseases; may not predict efficacy for other indications |
| Multivariate Analysis | Agglomerative cluster analysis of gene expression data (e.g., 5-gene subset for osteogenic potential) [93] | Accounts for multiple factors; potentially more predictive for complex functions | Technically challenging; requires specialized expertise and validation |
| Matrix-Based Approach (ISCT) | Gene/protein expression data coupled with functional assays using appropriate responder cells [93] | Comprehensive assessment; tailored to specific clinical indications | Complex implementation; high resource requirements |
Advanced Potency Assay Development: A Case Study
Recent advances in potency assay development address the need for greater physiological relevance and standardization. A robust example is an assay designed to measure the anti-inflammatory capacity of MSCs in M1 macrophage-driven diseases [98].
Experimental Protocol: Anti-inflammatory Potency Assay
Reagents and Equipment:
- THP-1 monocyte cell line
- Skin-derived ABCB5+ MSCs or other MSC populations
- Macrophage differentiation agents (PMA, IFN-γ, LPS)
- IL-1RA ELISA kit
- Flow cytometry equipment with CD36 and CD80 antibodies
- TNF-α detection assay
Methodology:
- Macrophage Differentiation and Polarization: Differentiate THP-1 monocytes into M1-polarized macrophages using phorbol 12-myristate 13-acetate (PMA), followed by stimulation with interferon-gamma (IFN-γ) and lipopolysaccharide (LPS) [98].
- Coculture Establishment: Coculture polarized M1 macrophages with MSCs at optimized ratios (determined through preliminary titration experiments).
- Marker Validation: Confirm successful macrophage differentiation and M1 polarization via flow cytometry analysis of CD36 and CD80 surface markers.
- Functional Confirmation: Verify M1 polarization through measurement of proinflammatory tumor necrosis factor alpha (TNF-α) release.
- Potency Measurement: Quantify interleukin-1 receptor antagonist (IL-1RA) secretion by MSCs in the coculture system using a validated ELISA.
- Data Analysis: Compare IL-1RA levels against predetermined specifications for batch release.
This assay exemplifies the necessary components of a modern potency test: physiological relevance, robust validation, and quantifiable endpoints correlated with proposed mechanisms of action.
Diagram 1: Anti-inflammatory potency assay workflow
Emerging Technologies and Novel Approaches
Innovative strategies are addressing the limitations of traditional potency assays by incorporating multidimensional assessment and mechanism-based prediction tools.
The CLIP Scale: A Hierarchical Framework for Efficacy Prediction
A novel approach to potency assessment involves the Clinical Indications Prediction (CLIP) scale, which predicts therapeutic efficacy based on TWIST1 expression levels in MSCs [93]. TWIST1 is a transcription factor that plays a prominent role in dictating MSC fate and function, mechanistically linking stem/progenitor and effector functions [93].
Experimental Protocol: Implementing the CLIP Scale
Reagents and Equipment:
- Human MSC isolates from relevant tissue sources
- RNA isolation kit
- qPCR equipment and reagents for TWIST1 quantification
- Colony-forming unit fibroblast (CFU-F) assay materials
- Functional assay reagents (angiogenic, immunomodulatory)
Methodology:
- TWIST1 Expression Analysis: Isolate RNA from MSC batches and quantify TWIST1 expression levels using qPCR.
- CFU-F Assay Correlation: Perform standard CFU-F assays to establish correlation between TWIST1 levels and progenitor activity.
- Functional Stratification: Stratify MSC batches based on TWIST1 expression:
- High TWIST1: Associated with rapid growth, high CFU-F activity, and pro-angiogenic phenotype
- Low TWIST1: Associated with anti-inflammatory and immunomodulatory phenotype
- Indication-Specific Assignment: Assign high-TWIST1 MSCs to indications requiring angiogenic potential (e.g., ischemic conditions) and low-TWIST1 MSCs to inflammatory conditions (e.g., autoimmune diseases).
- Validation: Confirm predictive value through in vivo models of specific diseases.
The CLIP scale represents a significant advancement as it predicts differences in growth, survival, stem/progenitor, and effector functions rather than focusing on a single parameter [93].
MSC Polarization for Enhanced Predictability
Another emerging approach involves preconditioning MSCs to enhance specific therapeutic functions:
Experimental Protocol: TLR Priming of MSCs
Reagents:
- Toll-like Receptor (TLR) agonists (TLR4 agonist: LPS; TLR3 agonist: Poly I:C)
- Standard MSC culture materials
- Functional assay reagents
Methodology:
- TLR Priming: Prime MSCs with TLR4 agonists (MSC1 phenotype) or TLR3 agonists (MSC2 phenotype) [93].
- Phenotype Validation:
- MSC1: Characterize proinflammatory mediator expression
- MSC2: Characterize immunosuppressive mediator expression
- Functional Testing: Assess immunomodulatory capacity in mixed lymphocyte reactions or other relevant functional assays.
- Correlation with Efficacy: Evaluate enhanced therapeutic potential in disease-specific models.
This polarization approach demonstrates how preconditioning can reduce heterogeneity and enhance predictability of MSC therapeutic effects.
The Scientist's Toolkit: Essential Research Reagents
Implementing robust potency assays requires specific reagents and tools standardized across the research community.
Table 3: Essential Research Reagents for MSC Potency Assessment
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Surface Marker Antibodies | CD73, CD90, CD105, CD14, CD34, CD45, HLA-DR [2] | MSC characterization per ISCT criteria | Lot-to-lot variability; validation required for consistency |
| Differentiation Kits | Osteogenic: Dexamethasone, β-glycerophosphate, ascorbate-2-phosphate [2] | Tri-lineage differentiation potential assessment | Supplier variations may affect differentiation efficiency |
| Cytokine/Chemokine Detection | ELISA kits for CXCL5, IL-8, VEGF, IL-1RA, TSG-6 [93] [98] | Quantification of secreted factors as potency markers | Establish validated reference ranges for each analyte |
| Cell Culture Supplements | Fetal Bovine Serum, platelet lysates, defined growth factors [95] | Maintenance of MSC properties during expansion | Significant impact on MSC characteristics; require rigorous qualification |
| Functional Assay Systems | THP-1 monocyte line, peripheral blood mononuclear cells (PBMCs), endothelial tube formation assays [93] [98] | Assessment of immunomodulatory and angiogenic potency | Donor variability in primary cells; use of standardized cell lines recommended |
Signaling Pathways Relevant to MSC Potency
Understanding the molecular mechanisms governing MSC functions provides the foundation for mechanism-based potency assays. Several key pathways regulate MSC therapeutic properties, particularly their immunomodulatory and differentiation capacities.
Diagram 2: Signaling pathways in MSC functional phenotypes
The diagram illustrates two major phenotypic directions for MSCs regulated by key signaling molecules. The immunomodulatory phenotype, characterized by TWIST1 downregulation, can be induced by IFN-γ and TNF-α exposure, resulting in expression of immunosuppressive factors including IDO, CD274/PD-L1, IL-4, and IL-10 [93]. In contrast, high TWIST1 expression maintains a pro-angiogenic phenotype associated with secretion of VEGF, CXCL5, and IL-8 [93]. These distinct signaling pathways provide potential targets for potency assay development and MSC product characterization.
Standardization Challenges and Future Directions
Despite advances in potency assay development, significant challenges remain in standardizing these approaches across the MSC field.
Key Standardization Barriers
- Heterogeneity of MSC Sources: Different tissue sources (bone marrow, adipose, umbilical cord) yield MSCs with distinct functional properties, making universal standardization challenging [94] [95].
- Manufacturing Process Variations: Differences in isolation techniques, culture media, supplements, and passage methods significantly impact MSC characteristics and potency [94] [96].
- Lack of Reference Materials: The absence of universally accepted reference MSC lines or standardized reagents contributes to inter-laboratory variability [94].
- Disease-Specific Potency Requirements: Different clinical indications may require distinct MSC functions, necessitating multiple potency assays for the same product [93].
Implementing a Standardization Framework
A multi-stakeholder approach is essential for advancing MSC standardization:
- Consensus Building: Engage researchers, clinicians, industry partners, and regulatory agencies to develop disease-specific potency assay recommendations.
- Reference Material Development: Establish well-characterized MSC reference materials for assay calibration and comparison.
- Process Control Guidelines: Define critical process parameters that significantly impact MSC potency and require tight control.
- Advanced Analytics: Implement multi-omics approaches (transcriptomics, proteomics, secretomics) to develop comprehensive potency signatures rather than relying on single parameters.
Recent stakeholder analyses indicate strong support for developing standardized assays that enable comparison across manufacturers, processes, and cell sources, though concerns remain about standardizing the cells themselves [94]. A unified cross-stakeholder approach will help advance MSC therapeutics and facilitate their clinical translation [94].
The development of robust, predictive potency assays represents the most critical unmet need in the clinical translation of MSC-based therapies. While significant challenges remain due to the inherent heterogeneity of MSC populations and the multifactorial nature of their therapeutic mechanisms, substantial progress is being made. Approaches such as the CLIP scale, mechanism-based functional assays, and TLR priming protocols provide promising pathways toward greater predictability and standardization.
The future of MSC potency assessment likely lies in multivariate approaches that integrate measurements of specific secretory profiles, functional responses to inflammatory cues, and molecular signatures predictive of therapeutic efficacy. As these tools evolve and gain standardization across the field, they will enable more reliable manufacturing of MSC products with predictable clinical outcomes, ultimately fulfilling the promise of MSC-based therapies for a broad range of human diseases.
Head-to-Head: A Data-Driven Comparison of BM-MSC and AD-MSC Efficacy
Within regenerative medicine and advanced therapy medicinal product (ATMP) development, the selection of an optimal mesenchymal stem cell (MSC) source is a critical foundational decision. The functional utility of MSCs is inherently constrained by their ex vivo expansion potential and innate proliferative rate, which directly impacts the feasibility of generating clinically relevant cell numbers. This technical analysis provides a comparative evaluation of the proliferation and clonogenic capacity of MSCs derived from prominent somatic and perinatal sources, specifically bone marrow (BM-MSCs), adipose tissue (AT-MSCs), and umbilical cord (UC-MSCs). The objective is to deliver a data-driven framework to assist researchers and drug development professionals in selecting the most appropriate cell source for their specific therapeutic applications, with a focus on expansion efficiency.
Quantitative Comparison of Proliferation and Clonogenic Potential
A direct comparison of MSC sources reveals clear hierarchical differences in their growth characteristics and isolation success. The data below, synthesized from multiple comparative studies, provides a quantitative foundation for source selection.
Table 1: Comparative Analysis of Proliferation and Clonogenic Capacity of Different MSC Sources
| MSC Source | Proliferation Potential | Colony Frequency (CFU-F) | Isolation Success Rate | Reported Population Doubling Time | Senescence Tendency |
|---|---|---|---|---|---|
| Bone Marrow (BM-MSC) | Low (Reference) [99] | Lower than AT-MSC [99] | Moderate [99] | ~30-40 hours (Varies with donor age) [3] | Higher [99] |
| Adipose Tissue (AT-MSC) | Intermediate (Faster than BM-MSC) [99] | Higher than BM-MSC [99] | High [99] | Shorter than BM-MSC [3] | Intermediate [99] |
| Umbilical Cord (UC-MSC) | Highest (Faster than AT & BM) [99] [3] | Lower than AT-MSC [99] | Lower than BM and AT [99] | ~20 hours (e.g., Menstrual Blood SC) [3] | Lowest [99] |
Table 2: Functional and Practical Considerations for MSC Source Selection
| MSC Source | Key Advantages for Expansion | Primary Limitations | Ideal Use-Case Scenarios |
|---|---|---|---|
| Bone Marrow (BM-MSC) | Most established, gold-standard history [100] | Invasive harvesting; low yield; donor age-dependent decline [3] [100] | Autologous therapy where UC/AT not suitable; hematopoiesis support studies [99] |
| Adipose Tissue (AT-MSC) | High initial yield (~1B cells/300g tissue); less invasive harvest [3] [100] | Donor metabolic health may influence function [6] | High-cell-number autologous therapies (e.g., reconstructive surgery) [3] [100] |
| Umbilical Cord (UC-MSC) | High proliferative rate; low immunogenicity; "young" phenotype [3] [99] | Finite source (birth); allogeneic by nature [100] | Allogeneic "off-the-shelf" ATMPs; large-scale manufacturing [3] |
Experimental Protocols for Assessing Proliferation and Clonogenicity
Standardized, robust assays are essential for quantifying the parameters discussed above. The following section details key methodologies cited in the literature.
Colony-Forming Unit Fibroblast (CFU-F) Assay
The CFU-F assay is the gold-standard method for quantifying the clonogenic potential, and thus the stemness, of a primary MSC population [9].
- Objective: To determine the frequency of precursor cells within a heterogeneous isolate that are capable of proliferation and formation of a discrete colony.
- Procedure:
- Plating: Primary cells are plated at a very low density (e.g., 10-100 cells/cm²) in multi-well plates or dishes to ensure isolated growth [9].
- Culture: Cells are cultured in a standard MSC growth medium, such as αMEM supplemented with 5-10% human platelet lysate (hPL) or fetal bovine serum (FBS), and 1% penicillin/streptomycin for 1-3 weeks without disturbance [6] [9].
- Fixation and Staining: After sufficient time for colony formation, the culture medium is removed. Cells are washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde or 100% methanol, and stained with crystal violet or Giemsa stain for visualization [9].
- Quantification: Only colonies containing more than 50 cells are counted as a CFU-F. The frequency is calculated as: (Number of Colonies Formed / Number of Cells Seeded) × 100% [9].
Population Doubling and Growth Kinetics Assay
This assay provides direct measurement of proliferation rate over multiple passages, offering critical data for planning manufacturing scale-up.
- Objective: To track the expansion potential and growth kinetics of MSCs during serial passaging.
- Procedure:
- Seeding and Harvesting: A known number of cells (e.g., 1x10³ cells/cm²) is seeded in culture flasks. Upon reaching 80-90% confluence, cells are detached using a protease like trypsin or TrypLE Select [6].
- Cell Counting: The total number of viable cells is counted using an automated cell counter or a hemocytometer with trypan blue exclusion at the end of each passage.
- Calculation: The population doubling (PD) for each passage is calculated using the formula: PD = Log₂(Nᵢ / N₀), where Nᵢ is the cell yield at harvest and N₀ is the initial number of cells seeded. The cumulative PDs are summed over all passages to assess long-term expansion potential [75].
- Proliferation Assays: Tools like the Cell Counting Kit-8 (CCK-8) can be used for more frequent, non-destructive monitoring of proliferation within a passage. This kit utilizes a tetrazolium salt that is reduced by cellular dehydrogenases to a water-soluble formazan dye, the absorbance of which correlates with the number of metabolically active cells [6].
Molecular Regulation of MSC Stemness and Proliferation
The proliferative and clonogenic capacities of MSCs are intrinsically regulated by a network of transcription factors and signaling pathways that maintain their undifferentiated state. Understanding this molecular basis is key to optimizing culture conditions and preserving functionality during expansion.
Diagram 1: Molecular regulation of MSC stemness. Key transcription factors like TWIST1/2, OCT4, SOX2, and HOXB7 promote proliferation and maintain stemness by epigenetically suppressing senescence genes such as p16 and p21 [75].
The following workflow integrates molecular knowledge with practical cell culture, outlining a typical experiment from tissue isolation to functional validation of MSC quality after expansion.
Diagram 2: Experimental workflow for MSC isolation and qualification. The process from tissue harvest to characterization involves critical quality control checkpoints to ensure expanded cells retain desired properties [9].
The Scientist's Toolkit: Essential Reagents and Materials
Successful isolation and expansion of MSCs require a carefully selected set of reagents and tools. The following table details essential components for related experiments.
Table 3: Key Research Reagent Solutions for MSC Isolation and Expansion
| Reagent/Material | Function/Application | Example & Notes |
|---|---|---|
| Enzymatic Digestion Cocktail | Liberates stromal vascular fraction (SVF) from tissue matrix. | Collagenase (e.g., Type I or II); often used at 0.1% concentration for adipose tissue [9] [100]. |
| Culture Medium | Provides nutrients and growth factors for MSC expansion. | αMEM or DMEM, supplemented with 5-10% Human Platelet Lysate (hPL) or FBS, and 1% Penicillin/Streptomycin [6] [9]. |
| Cell Detachment Reagent | Passaging and harvesting adherent MSCs. | Trypsin-EDTA or recombinant enzymes like TrypLE Select, which is less aggressive [6]. |
| Proliferation Assay Kit | Quantifies metabolically active cells; tracks growth. | Cell Counting Kit-8 (CCK-8), which uses a water-soluble tetrazolium salt [6]. |
| Flow Cytometry Antibodies | Confirms MSC identity per ISCT criteria. | Fluorochrome-conjugated antibodies against CD73, CD90, CD105 (positive) and CD45, CD34, HLA-DR (negative) [6] [3] [9]. |
| Cryopreservation Medium | Long-term storage of MSC stocks. | Culture medium supplemented with 10% DMSO and a higher concentration of serum or hPL [9]. |
The choice of MSC source presents a clear trade-off between well-characterized but limited somatic sources and highly potent perinatal ones. UC-MSCs consistently demonstrate superior proliferation rates and lower senescence, making them a compelling candidate for allogeneic ATMPs requiring large-scale expansion. Conversely, AT-MSCs offer a robust balance of good proliferative capacity and high initial yield, favoring autologous applications where less invasive harvesting is paramount. While BM-MSCs remain a valuable benchmark, their slower growth and donor-dependent variability can be limiting for industrial-scale production. Future research should focus on standardizing isolation and culture protocols across laboratories and further elucidating the molecular mechanisms that can be harnessed to sustainably enhance the stemness of all MSC sources during ex vivo manipulation.
Mesenchymal stem cells (MSCs) represent a cornerstone of regenerative medicine due to their capacity for self-renewal and multilineage differentiation into mesodermal lineages including osteogenic, chondrogenic, and adipogenic pathways [51]. The therapeutic application of MSCs in tissue engineering and regenerative medicine (TERM) relies heavily on understanding the factors that govern their lineage commitment and differentiation potential [101]. While MSCs can be isolated from various tissues, those derived from bone marrow and adipose tissue remain the most extensively studied and clinically utilized sources, particularly for autologous cell-based therapies [102]. The differentiation processes are complex, tightly regulated events controlled by specific signaling cascades, transcription factors, and epigenetic mechanisms [51] [103].
A critical aspect of MSC biology is the growing evidence that the tissue source significantly influences their differentiation propensity—a concept fundamental to selecting the optimal cell source for specific clinical applications [102] [101]. Bone marrow-derived MSCs (BM-MSCs) and adipose tissue-derived MSCs (ATSCs), while sharing similar surface marker profiles, exhibit distinct and often reciprocal differentiation biases [102]. Understanding these intrinsic differences, along with the molecular pathways that govern lineage specification, is essential for advancing targeted regenerative strategies for conditions ranging from bone fractures and osteoarthritis to soft tissue defects [51] [101] [103]. This review provides a comparative analysis of the osteogenic, chondrogenic, and adipogenic potential of MSCs from different sources, with a focus on the underlying molecular mechanisms and experimental methodologies.
Molecular Regulation of Trilineage Differentiation
The commitment of MSCs to a specific lineage is orchestrated by a network of key transcription factors and signaling pathways. These molecular regulators often operate in a mutually exclusive manner, where the activation of one lineage program simultaneously suppresses alternative fates [51].
Osteogenic Differentiation
The Runx2 transcription factor is the master regulator of osteoblast commitment [103]. Studies on Runx2-deficient mice demonstrate a complete absence of osteoblasts and bone formation, establishing its pivotal role [103]. Runx2 activation in human BM-MSCs induces the expression of early osteogenic markers, including collagen I, alkaline phosphatase, and osteocalcin [103]. The Wnt/β-catenin signaling pathway also plays a critical role in promoting osteoblastogenesis [103]. Additionally, the transcription factor Osterix (Osx) acts downstream of Runx2, and its deficiency also results in a lack of osteoblast formation [103]. Signaling through bone morphogenetic proteins (BMPs), particularly BMP-2, promotes osteogenesis by enhancing Runx2 and Osx expression [103].
Chondrogenic Differentiation
Chondrogenic differentiation is primarily regulated by the SRY-related HMG-box transcription factor 9 (SOX9), which is essential for chondrocyte differentiation and cartilage formation [103]. This process is typically induced in vitro by members of the transforming growth factor-beta (TGF-β) superfamily, especially TGF-β1 and TGF-β3 [101] [104] [102]. These factors stimulate the production of a characteristic extracellular matrix rich in type II collagen and aggrecan [101] [104]. A critical consideration in chondrogenic differentiation is the avoidance of hypertrophy, often marked by type X collagen expression, which can lead to undesirable endochondral ossification [104].
Adipogenic Differentiation
Adipogenesis is coordinated by a cascade of transcription factors, with Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) serving as the central regulator [101]. The process occurs in two main phases: an early phase characterized by the upregulation of CCAAT/enhancer-binding proteins (CEBPB and CEBPD), and a late phase marked by the expression of PPARγ, CEBPA, and lipid-handling proteins like FABP-α and LPL [101]. The activation of adipogenic pathways often reciprocally inhibits osteogenic differentiation, highlighting the delicate balance between these lineages [51].
Table 1: Key Transcription Factors and Markers in MSC Differentiation
| Lineage | Master Regulator | Key Markers | Inductive Factors |
|---|---|---|---|
| Osteogenic | Runx2, Osterix | Alkaline Phosphatase (ALP), Osteocalcin, Collagen I, Mineralized nodules (Alizarin Red S+) | BMP-2, Dexamethasone, β-glycerophosphate, Ascorbic Acid [102] [103] |
| Chondrogenic | SOX9 | Aggrecan, Collagen II, Sox9, Proteoglycans (Alcian Blue+) | TGF-β1, TGF-β3, BMP-2, Dexamethasone, Ascorbate [101] [104] [102] |
| Adipogenic | PPARγ | FABP-α, LPL, Leptin, Lipid vacuoles (Oil Red O+) | Dexamethasone, IBMX, Indomethacin, Insulin [101] [102] |
Comparative Differentiation Potential by MSC Source
The tissue of origin is a major determinant of MSC differentiation capacity, imparting a kind of "epigenetic memory" that biases cells toward specific lineages [102].
Bone Marrow-Derived MSCs (BM-MSCs)
BM-MSCs are considered the gold standard for osteogenic differentiation [104] [102]. They demonstrate a stronger inherent potential for bone formation compared to ATSCs, characterized by higher expression of Runx2, earlier and greater alkaline phosphatase activity, and more robust calcium deposition [102]. This superior osteogenic capacity is linked to the hypomethylation of the Runx2 promoter in BM-MSCs, which facilitates its expression [102]. In chondrogenesis, BM-MSCs also show strong performance, with significant upregulation of aggrecan, SOX9, and type II collagen, though they may also exhibit higher levels of the hypertrophic marker type X collagen [104]. Conversely, BM-MSCs possess a relatively lower adipogenic potential, correlated with the hypermethylation of the PPARγ promoter [102].
Adipose Tissue-Derived MSCs (ATSCs)
ATSCs exhibit a reciprocal differentiation profile, showing a superior adipogenic potential compared to BM-MSCs [102]. They efficiently activate PPARγ and downstream adipogenic markers like adiponectin, LPL, and leptin, and form more lipid vesicles [101] [102]. This predisposition is underpinned by the hypomethylation of the PPARγ promoter in ATSCs [102]. While ATSCs can undergo osteogenic and chondrogenic differentiation, their potential is generally lower than that of BM-MSCs [102]. Their chondrogenic capacity may be limited by reduced expression of receptors for TGF-β and BMPs [101].
Synovial membrane and synovial fluid MSCs are of interest for cartilage repair due to their intra-articular origin. While they express standard MSC markers, their in vitro chondrogenic potential is not superior to that of age-matched BM-MSCs, though they may offer the advantage of lower hypertrophic gene expression [104]. Studies also indicate that even within a single tissue type, such as adipose tissue, the specific anatomical depot can influence the differentiation efficiency of the isolated ASCs [105].
Table 2: Comparative Differentiation Potential of MSCs from Different Sources
| Cell Source | Osteogenic Potential | Chondrogenic Potential | Adipogenic Potential | Key Molecular Characteristics |
|---|---|---|---|---|
| Bone Marrow (BM-MSCs) | High [104] [102] | High (may show hypertrophy) [104] | Low [102] | Runx2 promoter hypomethylation; PPARγ promoter hypermethylation [102] |
| Adipose Tissue (ATSCs) | Moderate [102] | Moderate [101] [102] | High [102] | PPARγ promoter hypomethylation; Runx2 promoter hypermethylation [102] |
| Synovial Fluid (SF-MSCs) | Not fully characterized | High (with low hypertrophy) [104] | Not fully characterized | CD44 positive; share common marker with chondrocytes [104] |
Experimental Protocols for Assessing Differentiation Potential
Standardized in vitro assays are critical for evaluating the trilineage differentiation potential of MSCs, a prerequisite for their characterization and therapeutic application.
Osteogenic Differentiation Protocol
- Cell Seeding: Plate MSCs at a density of 4 × 10³ cells/cm² in a multi-well plate and culture until confluence [102].
- Induction Medium: Use a basal medium (e.g., DMEM) supplemented with 10% FBS, 1 nM dexamethasone, 50 μM ascorbic acid, and 20 mM β-glycerophosphate [102].
- Culture Duration: Maintain cells in the induction medium for 14-21 days, refreshing the medium every 2-3 days.
- Analysis:
- Histological Staining: Fix cells with 70% ethanol and stain with Alizarin Red S (0.5-2%, pH 4.1-4.3) for 5-10 minutes to detect calcium deposits and mineralized nodules [102] [106].
- Gene Expression Analysis: Quantify mRNA levels of osteogenic markers like Alkaline Phosphatase (ALP), Osteocalcin, Bone Morphogenetic Protein 2 (BMP2), and BGLA using qRT-PCR [106].
Chondrogenic Differentiation Protocol
- Cell Preparation: Use a pellet or micromass culture system. For pellets, centrifuge 0.25-0.5 × 10⁶ MSCs in a conical tube to form a pellet [104] [102].
- Induction Medium: Use a serum-free basal medium supplemented with 10 ng/ml TGF-β3, 500 ng/ml BMP-2, 10⁻⁷ M dexamethasone, 50 μg/ml ascorbate-2-phosphate, 40 μg/ml proline, 100 μg/ml pyruvate, and 1% ITS+ Premix [104] [102].
- Culture Duration: Culture pellets for 21-28 days without disturbing, refreshing the medium every 2-3 days.
- Analysis:
- Histological Staining: Fix pellets in paraformaldehyde, embed in paraffin, section, and stain with Alcian blue (at pH 2.5) to detect sulfated proteoglycans in the extracellular matrix [104].
- Immunohistochemistry: Perform staining for type II collagen [104].
- Gene Expression Analysis: Analyze expression of SOX9, AGGREcan (ACAN), and type II collagen (COL2A1) via qRT-PCR [104] [106].
Adipogenic Differentiation Protocol
- Cell Seeding: Plate MSCs at 4 × 10³ cells/cm² and culture until post-confluence [102].
- Induction Medium: Use basal medium supplemented with 500 nM dexamethasone, 0.5 mM isobutylmethylxanthine (IBMX), 50 μM indomethacin, and 10 μg/ml insulin [102].
- Culture Duration: Maintain cells for 21 days, with medium changes every 3 days.
- Analysis:
Visualizing Signaling Pathways and Experimental Workflows
The following diagrams illustrate the core signaling pathways governing differentiation and a standard experimental workflow.
Signaling Pathways in MSC Differentiation
Trilineage Differentiation Experimental Workflow
The Scientist's Toolkit: Essential Reagents for Differentiation Studies
Successful differentiation experiments require a suite of specific reagents and growth factors. The following table details key components used in standard protocols.
Table 3: Essential Research Reagents for MSC Differentiation Studies
| Reagent Category | Specific Examples | Function in Differentiation |
|---|---|---|
| Induction Factors | Dexamethasone, BMP-2, BMP-6, TGF-β1, TGF-β3, bFGF, Insulin, IBMX, Indomethacin | Activate specific signaling pathways (e.g., BMP/Smad, TGF-β/Smad) and regulate master transcription factors to initiate and sustain lineage commitment [101] [104] [102]. |
| Media Supplements | Ascorbic Acid (or Ascorbate-2-phosphate), β-glycerophosphate, Sodium Pyruvate, Proline, ITS+ Premix (Insulin, Transferrin, Selenium) | Support extracellular matrix synthesis (e.g., collagen), provide an energy source, and enable mineralization in osteogenesis [104] [102]. |
| Staining Kits & Dyes | Alizarin Red S, Oil Red O, Alcian Blue, Safranin O | Histological detection of differentiation endpoints: calcium deposits (osteogenesis), lipid vacuoles (adipogenesis), and proteoglycans (chondrogenesis) [104] [102] [106]. |
| Antibodies | Anti-CD73, -CD90, -CD105, -CD34, -CD45, -CD44, -STRO-1; Anti-Collagen Type II, -Aggrecan | Flow cytometric immunophenotyping of MSC surface markers; immunohistochemical validation of differentiated cell phenotypes [51] [104] [106]. |
| Cell Culture Consumables | Collagenase Type I, Trypsin-EDTA, Ficoll-Paque, Low/High Glucose DMEM, Fetal Bovine Serum (FBS) | Enzymatic isolation of cells from tissues, cell passaging, and provision of basal culture environment for cell expansion and differentiation [102] [106] [105]. |
The lineage-specific differentiation potential of MSCs is intrinsically linked to their tissue of origin, with BM-MSCs exhibiting a clear bias toward osteogenesis and ATSCs toward adipogenesis, largely governed by epigenetic mechanisms such as the promoter methylation status of key genes like Runx2 and PPARγ [102]. Chondrogenic potential is robust in BM-MSCs, while synovial-derived cells may offer advantages for cartilage engineering due to reduced hypertrophy [104]. These inherent biases are critical considerations for designing effective cell-based therapies. The choice of MSC source should be strategically aligned with the target tissue for regeneration. Furthermore, standardizing differentiation protocols and analytical methods is paramount for generating comparable and reproducible data across studies. Future research delving deeper into the epigenetic landscape and the interplay of signaling pathways will further refine our ability to harness the full therapeutic potential of MSCs for regenerative medicine.
The therapeutic paradigm for Mesenchymal Stem Cells (MSCs) has shifted from a focus on their differentiation potential to recognizing their profound paracrine activity. The secretome—comprising the complete set of bioactive factors secreted by cells, including soluble proteins, cytokines, chemokines, growth factors, and extracellular vesicles (EVs) like exosomes—is now considered the primary mediator of their regenerative and immunomodulatory effects [107] [108]. This shift is supported by in vivo observations of transient and low engraftment rates of transplanted MSCs, despite demonstrating significant therapeutic benefits [107]. The MSC secretome exerts its functions by modulating the local cellular environment, promoting tissue repair, angiogenesis, and cell survival, while exerting potent anti-inflammatory and immunomodulatory effects [2]. This cell-free approach offers distinct advantages over whole-cell therapies, including reduced immunogenicity, minimal risk of tumorigenicity, and fewer ethical concerns, positioning it as a promising and innovative tool for regenerative medicine [109] [107].
The composition and potency of the secretome are not uniform but are dynamically influenced by the MSC tissue source, donor characteristics, and specific environmental cues encountered during culture [107]. This technical guide provides an in-depth analysis of the cytokine and growth factor profiles that define the immunomodulatory strength of secretomes derived from two of the most prevalent MSC sources: bone marrow (BM-MSCs) and adipose tissue (AD-MSCs), within the context of a broader thesis on MSC research.
Compositional Analysis of the MSC Secretome
The therapeutic efficacy of the MSC secretome is rooted in its complex molecular composition. A comprehensive understanding of its constituents is essential for rational therapeutic design and potency assessment.
Soluble Factors and Vesicular Cargo
The secretome encompasses two primary functional compartments: soluble factors and vesicular cargo. Soluble factors include a wide array of cytokines, chemokines, and growth factors that directly interact with recipient cells via surface receptors [108] [110]. Concurrently, extracellular vesicles (EVs), particularly exosomes, act as key delivery vehicles, encapsulating and protecting a diverse molecular payload—including proteins, lipids, messenger RNAs (mRNAs), and microRNAs (miRNAs)—and facilitating its transfer to target cells [109] [111]. This dual-mode of action allows for both rapid signaling and sustained reprogramming of recipient cells.
Table 1: Key Functional Categories of Bioactive Molecules in the MSC Secretome
| Functional Category | Key Representative Molecules | Primary Documented Functions |
|---|---|---|
| Immunomodulatory | TGF-β, IL-10, PGE2, IDO, HGF, TSG-6, HLA-G5 [112] [108] | Suppresses T-cell proliferation; promotes M2 macrophage polarization; induces Treg differentiation [113] [112] |
| Pro-angiogenic | VEGF, IGF-1, HGF, bFGF [108] | Stimulates endothelial cell proliferation and new blood vessel formation [109] [108] |
| Osteogenic/Chondrogenic | BMPs, OPG, IL-6, TGF-β [109] [110] | Promotes osteoblast and chondrocyte differentiation; supports bone matrix formation [109] |
| Anti-apoptotic | bFGF, TGF, GM-CSF [108] | Enhances cell survival and inhibits programmed cell death in injured tissues [108] |
| Trophic & Repair | HGF, TIMP1, IGF-1 [110] | Promotes tissue remodeling, wound healing, and regeneration [110] |
Quantitative Profiling of Secretome Components
Proteomic and bioinformatic analyses are critical for characterizing the secretome's quantitative profile. A systematic review found a substantial overlap in the protein compositions and functional annotations of BM-MSC and AD-MSC secretomes, suggesting convergent pathways for tissue regeneration [114]. For instance, gene ontology (GO) term analysis reveals enrichment for processes like "immune response" (GO:0006955) and "inflammatory response" (GO:0006954), driven by a core set of factors [110].
A targeted analysis of the Adipose-Derived Stem Cell (ADSC) secretome identified 107 key proteins [110]. STRING software analysis of these factors revealed their involvement in 844 enriched biological processes, which can be grouped into three primary therapeutic areas:
- Immunoregulation: 73 factors (e.g., TGFB1, IL10, CSF2) [110]
- Bone Regeneration: 13 factors (e.g., OPG, IL6) [110]
- Wound Healing and Soft Tissue Regeneration: 27 factors (e.g., HGF, TIMP1) [110]
Table 2: Select Quantified Factors in ADSC Secretome and Their Putative Roles
| Factor | Full Name | Putative Role in Oral and Maxillofacial Medicine | Key References |
|---|---|---|---|
| TGFB1 | Transforming Growth Factor Beta 1 | Immunomodulation; tissue repair; bone regeneration | [110] |
| IL10 | Interleukin 10 | Potent anti-inflammatory cytokine; immune suppression | [110] |
| CSF2 | Colony Stimulating Factor 2 (GM-CSF) | Macrophage activation; antimicrobial response | [110] |
| OPG | Osteoprotegerin | Inhibition of osteoclastogenesis; bone protection | [110] |
| HGF | Hepatocyte Growth Factor | Mitogenic for epithelial cells; wound healing; angiogenesis | [110] |
| TIMP1 | TIMP Metallopeptidase Inhibitor 1 | Inhibition of matrix metalloproteinases (MMPs); tissue remodeling | [110] |
Beyond proteins, the RNA content of secretomes, particularly within exosomes, is functionally significant. Sequencing of MSC-exosomes shows that their small RNAome is dominated by miRNAs and snoRNAs, but also reveals selective enrichment of specific miRNAs and tRNA halves, which may be related to the tissue origin and differentiation status of the parent MSCs [111].
The tissue origin of MSCs is a critical variable that shapes the secretory profile and consequent therapeutic potential.
Bone Marrow vs. Adipose Tissue MSC Secretome
BM-MSCs were the first discovered and remain the most extensively studied type. Their secretome is characterized by a rich profile of factors supporting osteogenesis and hematopoiesis [2]. AD-MSCs, derived from a more accessible and abundant tissue source, offer a higher yield of cells and possess a secretome with strong angiogenic and wound-healing properties [115] [110].
A systematic bioinformatics review indicates that while source-specific differences exist, there is a substantial overlap in the functional annotations and protein compositions of BM-MSC and AD-MSC secretomes, particularly concerning proteins involved in bone formation [114]. This suggests that for broad regenerative applications, the source may be less influential than previously thought. However, proteomic comparisons indicate that secretomes from induced pluripotent stem cell-derived MSCs (iMSCs) and umbilical cord-derived MSCs (UC-MSCs) are enriched with factors associated with proliferative potential and telomere maintenance. In contrast, secretomes from adult-tissue-derived BM-MSCs and AD-MSCs contain higher levels of fibrotic and extracellular matrix (ECM) proteins [109].
Influence of Cell Source on Experimental and Therapeutic Outcomes
The biological differences stemming from the cell source can directly influence experimental readouts and therapeutic efficacy. For example, the specific sorting of evolutionary conserved tRNA species into exosomes differs between AD-MSCs and BM-MSCs, an effect linked to the differentiation status and stemness of the cells as defined by Sox2, POU5F1A/B, and Nanog expression [111].
Furthermore, source impacts practical therapeutic aspects. UC-MSCs, for instance, are noted for their non-invasive collection, high proliferation rates, and strong immunomodulatory properties, making them attractive for allogeneic therapies [2] [108]. The choice of MSC source must therefore align with the specific therapeutic goal, whether it is bone regeneration (where BM-MSCs may be preferred), wound healing (where AD-MSCs may be superior), or treating immune-related disorders (where UC-MSCs show great promise).
Methodologies for Secretome Analysis and Functional Characterization
A robust analytical framework is essential to deconvolute the complex composition and function of the MSC secretome.
Standardized Protocol for Secretome Collection and Exosome Isolation
To ensure reproducible and contaminant-free secretome samples, standardized protocols are critical. The following workflow, adapted from current research, outlines a typical procedure for generating conditioned medium (CM) and isolating exosomes [111]:
Workflow:
- Cell Culture: Expand early-passage MSCs (e.g., P2-P3) in standard media supplemented with Fetal Bovine Serum (FBS).
- Preconditioning & Serum Starvation: Once cultures reach 70-80% confluence, replace the standard medium with a serum-free medium or medium containing exosome-depleted FBS (prepared by ultracentrifugation at 70,000 × g overnight) [111]. Culture for 24-48 hours.
- Collection of Conditioned Medium (CM): Collect the CM and subject it to a series of centrifugation steps to remove cells and debris:
- 500 × g for 10 minutes (twice)
- 2,000 × g for 15 minutes (twice)
- 10,000 × g for 30 minutes (twice)
- Exosome Isolation via Ultracentrifugation: Transfer the supernatant to Ultra-Clear tubes and ultracentrifuge at 70,000 × g for 1 hour at 4°C (e.g., using a SW32Ti rotor) [111]. Resuspend the resulting exosome pellet in PBS for immediate use or storage at -80°C.
Analytical Techniques for Secretome Characterization
Comprehensive secretome analysis requires a multi-modal approach:
- Protein/RNA Array Kits: Antibody-based arrays allow for the simultaneous quantification of dozens to hundreds of pre-selected proteins (cytokines, growth factors) from small sample volumes, providing a medium-throughput screening method [110].
- Small RNA-Sequencing: Next-generation sequencing of the RNA cargo of exosomes provides an unbiased, comprehensive view of the miRNA, snoRNA, and tRNA landscape. This requires a dedicated bioinformatics pipeline for adapter trimming, alignment, and differential expression analysis [111].
- Proteomic Analysis (Mass Spectrometry): Liquid chromatography-tandem mass spectrometry (LC-MS/MS) enables the unbiased identification and quantification of thousands of proteins in a secretome sample, forming the basis for systems biology approaches [114].
- Bioinformatics and Functional Enrichment Analysis: Tools like the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) are used to analyze protein-protein interaction networks and identify over-represented Gene Ontology (GO) terms and KEGG pathways, linking the molecular components to biological functions [114] [110].
Functional Assays for Immunomodulatory Strength
In vitro functional assays are indispensable for validating the biological activity predicted by compositional analysis.
- T-cell Proliferation Assay: Co-culture activated T-cells (e.g., from peripheral blood mononuclear cells - PBMCs) with MSC secretome or isolated exosomes. T-cell proliferation is measured using CFSE dilution or EdU incorporation via flow cytometry. A reduction in proliferation indicates immunosuppressive capacity [112].
- Macrophage Polarization Assay: Differentiate monocytes into M0 macrophages, then treat with MSC secretome in the presence of M1-inducers (e.g., LPS + IFN-γ). Assess polarization by measuring the expression of M1 (CD80, iNOS) and M2 (CD206, Arg1) markers using flow cytometry and qPCR, demonstrating a shift from pro-inflammatory M1 to anti-inflammatory M2 phenotypes [112].
- Treg Differentiation Assay: Isolate naïve CD4+ T-cells and stimulate them under Treg-polarizing conditions in the presence of the MSC secretome. The percentage of induced CD4+CD25+FOXP3+ regulatory T cells (Tregs) is quantified by flow cytometry, confirming the promotion of an immunotolerant environment [112].
Signaling Pathways Underlying Immunomodulation
The immunomodulatory strength of the MSC secretome is mediated through the orchestrated activation of several key signaling pathways in target immune cells.
Key Immunomodulatory Pathways
- TGF-β/Smad Pathway: Secreted TGF-β binds to its receptor (TGF-βRII/TGF-βRI complex), leading to the phosphorylation of Smad2/3. This complex partners with Smad4, translocates to the nucleus, and promotes the transcription of FoxP3, the master regulator of Treg differentiation, thereby fostering immune tolerance [112].
- PD-1/PD-L1 Axis: MSCs express PD-L1 on their surface, either directly or via exosomes. Upon interaction with PD-1 on activated T-cells, it recruits phosphatases SHP-1/SHP-2, which dephosphorylate key signaling molecules like ZAP70. This inhibits T-cell receptor (TCR) signaling, leading to cell cycle arrest in G0/G1 and suppression of T-cell proliferation [112].
- TSG-6 Mediated Macrophage Repolarization: In response to inflammatory signals like TNF, MSCs secrete TNF-α-stimulated gene 6 (TSG-6). TSG-6 acts on resident macrophages, reducing NF-κB signaling, which in turn dampens pro-inflammatory cytokine production and promotes a transition from an M1 to an M2 phenotype [113] [112].
Table 3: Key Research Reagent Solutions for Secretome Analysis
| Reagent / Kit | Function / Application | Key Considerations |
|---|---|---|
| Exosome-Depleted FBS | Serum supplement for cell culture during secretome production to minimize contaminating bovine exosomes. | Essential for preparing clean, well-defined conditioned medium. Prepared by overnight ultracentrifugation (e.g., 70,000 × g) [111]. |
| Proteome Profiler Array Kits | Simultaneous quantification of multiple cytokines, chemokines, and growth factors from secretome samples. | Ideal for targeted, medium-throughput screening. Provides semi-quantitative data on a predefined set of analytes [110]. |
| Small RNA-Sequencing Kits | Comprehensive, unbiased profiling of the small RNA content (miRNA, tRNA, snoRNA) within exosomes. | Requires specialized bioinformatics support for data analysis. Reveals selective enrichment of RNA species [111]. |
| Ultracentrifugation Equipment | Gold-standard method for isolating exosomes from conditioned medium based on size and density. | Requires high-cost instrumentation (e.g., SW32Ti rotor). Critical for obtaining pure exosome fractions for downstream analysis [111]. |
| Flow Cytometry Antibodies | Characterization of MSC surface markers (CD73, CD90, CD105) and analysis of immune cell phenotypes in functional assays. | For MSCs: Confirm expression of CD73, CD90, CD105 (≥95%) and lack of CD34, CD45, CD14, CD19, HLA-DR (≤2%) [2] [112]. For functional assays: Antibodies for T-cell (CD3, CD4, CD8), Treg (CD25, FOXP3), and macrophage (CD80, CD206) markers. |
The analysis of the MSC secretome represents a frontier in regenerative medicine, offering a cell-free therapeutic paradigm with significant potential for treating immune-mediated and degenerative diseases. This guide has detailed the core components, analytical methods, and functional mechanisms that define the immunomodulatory strength of secretomes from key MSC sources like bone marrow and adipose tissue. The field is rapidly advancing towards engineered solutions, such as preconditioning MSCs with inflammatory cytokines to enhance the immunomodulatory content of their exosomes [109], and using 3D culturing to better mimic the native cellular microenvironment and boost secretory profiles [107]. As research progresses, the future of secretome-based therapies lies in the development of standardized, potent, and potentially engineered "designer secretomes" tailored to specific clinical applications, ultimately unlocking their full potential to transform the treatment of human diseases.
The selection of an appropriate mesenchymal stem cell (MSC) source represents a pivotal decision point in developing effective regenerative therapies. While MSCs isolated from bone marrow (BM-MSCs) and adipose tissue (AT-MSCs) share fundamental characteristics—adherence to plastic, specific surface marker expression, and multipotent differentiation capacity—a growing body of evidence demonstrates that their biological properties and therapeutic potentials are far from identical [2]. These differences are not merely academic but have direct implications for clinical outcomes across various medical applications. The tissue-specific differentiation capacities, proliferative potentials, immunomodulatory strengths, and secretome profiles of MSCs vary significantly depending on their anatomical origin [33] [20] [116]. This technical guide provides an evidence-based framework for researchers and therapy developers to systematically select the optimal MSC source for orthopedic, immunologic, and soft tissue regeneration applications, drawing upon direct comparative studies and mechanistic insights into MSC biology.
Biological Characteristics: A Comparative Analysis of BM-MSCs and AT-MSCs
Phenotypic and Functional Differences
Despite meeting the International Society for Cellular Therapy (ISCT) minimal criteria for MSCs, BM-MSCs and AT-MSCs exhibit distinct biological profiles that inform their therapeutic specialization. Both cell types display similar fibroblast-like morphology and standard surface marker expression (CD73, CD90, CD105) while lacking hematopoietic markers [33] [20]. However, significant differences emerge in finer characterization, with AT-MSCs typically showing high expression of CD49d and low expression of Stro-1 compared to BM-MSCs [20].
Functionally, AT-MSCs demonstrate significantly greater proliferative capacity in culture, with shorter population doubling times and enhanced clonogenicity compared to BM-MSCs [33] [117] [20]. This expansion advantage makes AT-MSCs particularly attractive for applications requiring substantial cell numbers. Conversely, BM-MSCs exhibit superior differentiation capacity toward osteogenic and chondrogenic lineages, demonstrating higher alkaline phosphatase activity, mineralization potential, and expression of osteogenesis-related genes and proteins [33] [20]. Both MSC types show generally similar adipogenic differentiation potential, though some studies indicate AT-MSCs may have a slight advantage in lipid vesicle formation and adipogenesis-related gene expression [20].
Table 1: Comparative Biological Characteristics of BM-MSCs and AT-MSCs
| Characteristic | BM-MSCs | AT-MSCs | Significance |
|---|---|---|---|
| Proliferation Capacity | Moderate | High [33] [117] | AT-MSCs superior for rapid expansion |
| Osteogenic Potential | High [33] [20] | Moderate | BM-MSCs preferred for bone formation |
| Chondrogenic Potential | High [33] [20] | Moderate | BM-MSCs preferred for cartilage repair |
| Adipogenic Potential | Moderate | High [20] | AT-MSCs slightly superior for fat tissue formation |
| Immunomodulatory Effects | Moderate | Potent [33] | AT-MSCs show stronger immunosuppression |
| Colony-Forming Efficiency | Similar to AT-MSCs [33] | Similar to BM-MSCs [33] | Comparable self-renewal capacity |
| Harvesting Yield | Low (0.001-0.01% of nucleated cells) [20] | High (abundant from lipoaspirates) [20] | AT-MSCs more accessible in large quantities |
| Donor Age Sensitivity | High (declines with age) [39] | Lower | AT-MSCs more consistent across donor ages |
Secretome Profiles and Immunomodulatory Properties
The therapeutic effects of MSCs are increasingly attributed to their paracrine activity rather than direct differentiation, making the secretome composition a critical differentiator. Comparative analyses reveal distinct cytokine, growth factor, and chemokine secretion patterns between BM-MSCs and AT-MSCs [33]. AT-MSCs secrete higher levels of basic fibroblast growth factor (bFGF), interferon-γ (IFN-γ), and insulin-like growth factor-1 (IGF-1), contributing to their enhanced proliferative and immunomodulatory capacities [33]. In contrast, BM-MSCs produce more stem cell-derived factor-1 (SDF-1) and hepatocyte growth factor (HGF), factors associated with hematopoietic support and tissue repair [33].
These secretome differences translate to functional variation in immunomodulation. AT-MSCs demonstrate more potent immunomodulatory effects in comparative studies, showing enhanced capacity to suppress immune cell proliferation and modulate inflammatory responses [33]. This makes them particularly attractive for applications in autoimmune diseases, graft-versus-host disease, and other inflammatory conditions. The extracellular vesicles (EVs) and exosomes derived from both MSC types mirror these source-dependent functional variations, displaying distinct miRNA profiles and cargo compositions that influence their therapeutic effects [5] [118].
MSC Source Selection for Specific Therapeutic Applications
Orthopedic Applications: Bone and Cartilage Regeneration
For orthopedic applications involving bone and cartilage repair, the differentiation capacity of MSCs becomes the paramount selection criterion. BM-MSCs demonstrate superior osteogenic and chondrogenic potential in direct comparisons, making them the preferred choice for bone tissue engineering and cartilage regeneration [33] [20] [116]. Studies consistently show that BM-MSCs exhibit earlier and higher alkaline phosphatase activity, enhanced calcium deposition, and stronger expression of osteogenesis-related genes (osteopontin) and chondrogenesis-related markers compared to AT-MSCs [20].
The molecular basis for this superiority involves enhanced responsiveness to osteogenic and chondrogenic induction signals in BM-MSCs. Research indicates that BM-MSCs show differential expression of key regulatory genes in transforming growth factor-beta (TGF-β), Wnt, and fibroblast growth factor (FGF) signaling pathways when compared to AT-MSCs [119]. These pathway activations drive more robust extracellular matrix production and mineralization capacity essential for bone and cartilage formation. Additionally, BM-MSCs' native bone marrow environment may prime them for skeletal tissue regeneration through epigenetic programming and receptor expression profiles optimized for responding to skeletal repair signals.
Table 2: MSC Source Recommendations by Application Area
| Application Area | Recommended Source | Rationale | Key Supporting Evidence |
|---|---|---|---|
| Bone Regeneration | BM-MSCs [33] [117] [20] | Superior osteogenic differentiation & mineralization | Higher ALP activity, calcium deposition, osteogenic gene expression |
| Cartilage Repair | BM-MSCs [33] [20] [116] | Enhanced chondrogenic potential | Stronger proteoglycan production, chondrogenic gene expression |
| Immunomodulation | AT-MSCs [33] | More potent immunosuppressive effects | Greater T-cell suppression, anti-inflammatory cytokine secretion |
| Adipose Tissue Engineering | AT-MSCs [20] | Native predisposition to adipogenesis | Enhanced lipid vesicle formation, adipogenic gene expression |
| Wound Healing | AT-MSCs [117] | Strong pro-angiogenic secretome | Higher bFGF, VEGF secretion; enhanced proliferative capacity |
| Glioma Tropism | Either [39] | Similar migration to tumor sites | Comparable tropism with AT-MSCs offering harvest advantages |
Immunological Applications: Inflammation and Autoimmunity
For immunomodulatory applications, AT-MSCs demonstrate superior immunosuppressive capabilities compared to BM-MSCs [33]. This enhanced immunomodulation stems from both their secretome profile and direct cell-cell interaction capabilities. AT-MSCs secrete higher levels of factors like prostaglandin E2 (PGE2), which modulates macrophage polarization toward anti-inflammatory phenotypes and suppresses T-cell proliferation [2]. They also show enhanced expression of immunomodulatory surface markers that facilitate direct contact with immune cells.
The therapeutic implications of these differences are significant for conditions like graft-versus-host disease (GVHD), autoimmune disorders, and inflammatory conditions. In these contexts, the potent immunomodulation of AT-MSCs can more effectively suppress aberrant immune responses and resolve inflammation. Additionally, the easier harvest and greater expansion potential of AT-MSCs provide practical advantages for these applications, which often require substantial cell doses for therapeutic efficacy [20]. The emergence of MSC-derived extracellular vesicles as cell-free therapeutic alternatives further extends these applications, with source-dependent variations in EV composition influencing their immunomodulatory potency [118] [89].
Soft Tissue Regeneration and Other Applications
For soft tissue regeneration, particularly adipose tissue engineering, AT-MSCs demonstrate clear advantages due to their native tissue origin and enhanced adipogenic potential [20]. Studies show that AT-MSCs more readily undergo adipogenic differentiation with more abundant lipid vesicle formation and stronger expression of adipogenesis-related genes compared to BM-MSCs [20]. This makes them ideal for soft tissue reconstruction, breast reconstruction post-mastectomy, and treatment of soft tissue defects.
In neurological applications, particularly for glioma therapy where MSC tropism to tumors is utilized for targeted drug delivery, both MSC sources show similar migratory capacity toward glioma sites [39]. In this context, practical considerations favor AT-MSCs due to their easier harvest, greater expansion potential, and reduced donor age sensitivity [39]. For cardiovascular and angiogenic applications, the strong secretome of AT-MSCs rich in pro-angiogenic factors like VEGF and bFGF may provide advantages in promoting vascularization of ischemic tissues [33] [117].
Experimental Protocols for MSC Characterization
Standardized Isolation and Culture Methods
Proper isolation and culture techniques are essential for maintaining the inherent biological properties of different MSC sources. For BM-MSC isolation, bone marrow aspirates are typically collected with heparin anticoagulant, filtered through a 70μm strainer, and separated via density gradient centrifugation or direct plating [20] [116]. The mononuclear cell fraction is plated in culture vessels with Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) [20]. Non-adherent cells are removed after 24-48 hours, and adherent cells are maintained with medium changes twice weekly.
For AT-MSC isolation, adipose tissue is washed extensively with PBS, minced thoroughly, and digested with 0.1% collagenase type I for 30-60 minutes at 37°C with agitation [20]. The digestion is neutralized with culture medium, and the sample is centrifuged to separate the stromal vascular fraction (pellet) from adipocytes and debris. The pellet is resuspended, filtered through 100-70μm filters, and plated in culture vessels [20]. For clinical applications, human platelet lysate (hPL) represents a xenogeneic-free alternative to FBS that enhances MSC expansion while maintaining differentiation capacity and immunophenotype [33].
Differentiation Capacity Assessment
Osteogenic differentiation is induced by culturing MSCs to 70-80% confluence and switching to induction media containing DMEM, 10% FBS, 50μM ascorbate-2-phosphate, 10mM β-glycerophosphate, and 100nM dexamethasone [5] [20]. Cultures are maintained for 2-3 weeks with medium changes twice weekly. Successful differentiation is confirmed by alkaline phosphatase activity staining (day 7-10), mineralization detection through Alizarin Red S staining (day 21), and expression of osteogenic genes (RUNX2, osteocalcin) via RT-PCR.
Chondrogenic differentiation is typically performed using pellet culture systems. Approximately 2.5×10^5 MSCs are centrifuged to form a pellet cultured in chondrogenic media containing DMEM, 1% ITS+ premix, 100nM dexamethasone, 50μM ascorbate-2-phosphate, 40μg/mL proline, and 10ng/mL TGF-β3 [5]. After 3-4 weeks, pellets are assessed for proteoglycan deposition with Safranin O or Toluidine Blue staining, collagen type II formation by immunohistochemistry, and chondrogenic gene expression (SOX9, aggrecan, collagen type II).
Adipogenic differentiation is induced at cell confluence using media containing DMEM, 10% FBS, 0.5mM isobutylmethylxanthine, 1μM dexamethasone, 10μM insulin, and 200μM indomethacin [5] [20]. After 3 weeks, lipid accumulation is visualized by Oil Red O staining, and adipogenic gene expression (PPARγ, LPL, FABP4) is analyzed by RT-PCR.
Research Reagent Solutions for MSC Studies
Table 3: Essential Research Reagents for MSC Characterization
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Isolation Reagents | Collagenase Type I [20], Lymphoprep [33], Heparin [20] | Tissue dissociation, cell separation | Concentration and digestion time critical for viability |
| Culture Media | DMEM/F12 [39], αMEM [5], MSCGM [39] | Baseline cell culture & expansion | Serum source affects differentiation potential |
| Growth Supplements | FBS [20], hPL [33], GlutaMax [39] | Support proliferation & viability | hPL reduces xenogeneic risks for clinical applications |
| Differentiation Kits | StemPro Osteo/Chondro/Adipogenesis Kits [39] | Standardized lineage induction | Ensure lot-to-lot consistency for reproducibility |
| Characterization Antibodies | CD73, CD90, CD105 [20], CD34, CD45 [20] | Immunophenotyping by flow cytometry | Include appropriate isotype controls |
| Staining Reagents | Alizarin Red S [20], Oil Red O [20], Safranin O [5] | Detection of differentiation outcomes | Quantification methods available for mineralization |
| Growth Factors | TGF-β3 [5], FGF-2 [39], BMPs | Directed differentiation | Concentration and timing critical for efficacy |
Signaling Pathways Regulating MSC Behavior
The Krüppel-like factor 4 (KLF4) transcription factor serves as a critical regulator of MSC biology, with demonstrated roles in maintaining stemness properties and controlling differentiation initiation [119]. KLF4 is highly expressed in primitive MSC subtypes with enhanced clonal capacity and multilineage potential. During early differentiation induction, KLF4 expression decreases significantly within 24 hours, suggesting its role as a gatekeeper of the undifferentiated state [119].
Functional studies using knockdown approaches demonstrate that KLF4 depletion enhances both proliferation and early differentiation in MSCs, indicating its inhibitory function in these processes [119]. Mechanistically, KLF4 regulates the expression of key receptors in multiple signaling pathways: TGFBR1 (TGF-β signaling), FZD6 (Wnt signaling), and FGFR2 (FGF signaling) [119]. These pathways collectively influence MSC fate decisions, with TGF-β and Wnt signaling particularly important for chondrogenic and osteogenic differentiation, respectively, while FGF signaling promotes proliferative expansion. The coordinated regulation of these pathways by KLF4 positions it as a master regulator of the balance between self-renewal and lineage commitment in MSCs.
Clinical Translation Considerations
Practical and Regulatory Aspects
The translation of MSC-based therapies from research to clinical application requires careful consideration of practical and regulatory factors. Source selection impacts multiple aspects of clinical translation, including harvesting procedures, expansion capabilities, and safety profiles. BM-MSC harvesting is moderately invasive, requiring bone marrow aspiration from the iliac crest, with associated donor discomfort and morbidity [20]. In contrast, AT-MSCs can be obtained through minimally invasive liposuction procedures, typically yielding higher cell numbers from equivalent tissue volumes [20] [39].
The expansion potential of AT-MSCs provides practical advantages for clinical manufacturing, requiring fewer population doublings to achieve therapeutic doses. This is particularly relevant for autologous applications in older patients, where BM-MSCs show age-related declines in proliferation and differentiation capacity [39]. For allogeneic approaches, both MSC types exhibit low immunogenicity due to limited MHC class II expression, making them suitable for "off-the-shelf" therapies [2].
Emerging Trends: Extracellular Vesicles and Standardization
Cell-free therapies using MSC-derived extracellular vesicles (EVs) and exosomes represent a promising alternative to whole-cell transplantation, mitigating risks associated with direct cell administration [118] [89]. MSC-EVs retain therapeutic properties of their parent cells, including immunomodulation, pro-angiogenic effects, and tissue repair capabilities, while offering advantages in safety, stability, and storage [118]. Importantly, EVs maintain the source-dependent functional characteristics of their parent MSCs, making source selection equally relevant for EV-based therapies.
Current challenges in the field include lack of standardized protocols for EV isolation, characterization, and dosing [118] [89]. Clinical trials show substantial variation in these parameters, complicating cross-study comparisons and optimization of therapeutic regimens. Similarly, standardization in MSC culture conditions, differentiation protocols, and potency assays remains essential for advancing the field and achieving consistent clinical outcomes [33] [118].
The selection between BM-MSCs and AT-MSCs represents a critical determinant of therapeutic success in regenerative medicine applications. BM-MSCs demonstrate clear advantages for orthopedic applications requiring robust osteogenic and chondrogenic differentiation, while AT-MSCs excel in immunomodulatory contexts and scenarios requiring rapid expansion and abundant cell yields. The biological differences between these MSC sources—from their differentiation capacities and proliferation rates to their secretome profiles and immunomodulatory potencies—should be systematically evaluated against specific therapeutic requirements. As the field advances toward more refined applications and cell-free approaches, understanding these source-dependent variations will continue to inform the rational design of MSC-based therapies optimized for specific clinical indications.
The field of regenerative medicine is increasingly focusing on induced pluripotent stem cell-derived mesenchymal stem cells (iPSC-derived MSCs or iMSCs) as a solution to critical challenges hindering the commercialization of traditional MSC-based therapies. While primary MSCs from bone marrow and adipose tissue have demonstrated promising therapeutic potential, their clinical application is constrained by significant limitations, including donor-to-donor variability, limited expansion capacity, and product heterogeneity [120]. These factors create substantial barriers to manufacturing standardized, off-the-shelf cell therapies that meet rigorous regulatory requirements for consistency and potency.
The iPSC-derived MSCs represent a paradigm shift, offering a platform for producing uniform, scalable, and well-characterized cell populations. This whitepaper examines the key players driving innovation in this sector and analyzes the experimental protocols and standardized approaches that are positioning iMSCs as the future of commercial MSC-based regenerative medicine. By leveraging the unlimited self-renewal capacity of iPSCs, iMSCs overcome the fundamental supply chain and manufacturing challenges associated with primary MSCs, potentially enabling a new generation of standardized therapeutic products [120].
Key Players in the iPSC-Derived MSC Landscape
The commercialization landscape for iMSCs features established biotechnology companies and emerging specialists, all working to translate the technological advantages of iPSC-derived platforms into clinically viable and commercially successful products. These entities are developing proprietary technologies for the differentiation, scale-up, and characterization of iMSCs to ensure consistent product quality.
Table 1: Key Companies Developing iPSC-Derived MSC (iMSC) Therapeutics
| Company | Key Focus/Technology | Development Stage |
|---|---|---|
| Cynata Therapeutics | Pioneering iPSC-derived MSC production technologies [121] | Clinical development |
| Fujifilm CDI (Cellular Dynamics Inc) | Large-scale production of human cells from iPSCs [121] | Not specified |
| Eterna Therapeutics | mRNA-based cell engineering [121] | Not specified |
| Implant Therapeutics | iMSC development [121] | Not specified |
| Bone Therapeutics | Orthopedic applications [121] | Not specified |
| Brooklyn ImmunoTherapeutics | iMSC development [121] | Not specified |
| Citius Pharmaceuticals | iMSC development [121] | Not specified |
| Kiji Therapeutics | iMSC development [121] | Not specified |
The activity of these key players is set against a backdrop of vigorous clinical research. Globally, there are over 1,670 clinical trials involving MSCs registered on ClinicalTrials.gov [121]. Analysis of trial data reveals that nearly 75% of these studies aim to develop regenerative medicine products, while approximately 14% focus on disease modeling, and the remaining 11% are dedicated to drug discovery and cytotoxicity testing [121]. This robust clinical activity underscores the broad therapeutic potential of MSC-based approaches and highlights the need for the standardized cell sources that iMSCs provide.
The iMSC Advantage: Overcoming Primary MSC Limitations
Primary MSCs, whether sourced from bone marrow (BM-MSCs) or adipose tissue (AD-MSCs), present inherent biological and manufacturing challenges that complicate their commercial-scale production. These challenges include:
- Donor Heterogeneity: The biological characteristics and therapeutic potency of primary MSCs vary significantly based on donor age, health status, and tissue source [120].
- Limited Expansion Capacity: Primary MSCs undergo senescence after a limited number of population doublings in vitro, restricting the cell numbers obtainable from a single donation [120].
- Tissue Harvesting Invasiveness: Bone marrow aspiration is an invasive procedure for donors, and the yield of MSCs from bone marrow is substantially lower (approximately 500-fold less) than from adipose tissue [120].
iMSCs directly address these limitations. They offer a virtually unlimited cell source due to the indefinite self-renewal capacity of the parent iPSC line. This enables the creation of extensive Master Cell Banks (MCBs) and Working Cell Banks (WCBs), ensuring a consistent, well-characterized, and reproducible starting material for therapeutic production [120]. Furthermore, iMSCs can be generated from iPSCs derived from any donor, including patients with specific diseases, facilitating the development of both allogeneic ("off-the-shelf") and personalized autologous therapies.
Table 2: Functional Comparison of Primary MSCs vs. iMSCs
| Characteristic | Primary MSCs (BM/Adipose) | iPSC-Derived MSCs (iMSCs) |
|---|---|---|
| Scalability | Limited by donor tissue availability and senescence [120] | Essentially unlimited via iPSC self-renewal [120] |
| Uniformity | High donor-to-donor and batch-to-batch variability [120] | High consistency from defined iPSC banks [120] |
| Proliferative Capacity | Declines with age and in-vitro passaging [120] | Robust and sustained proliferation potential [120] |
| Manufacturing | Complex logistics for tissue sourcing | Streamlined, bank-based production system [120] |
| Genetic Stability | Risk of senescence during expansion [121] | Controlled differentiation reduces culture-induced changes |
Experimental Protocols for iMSC Generation and Characterization
Standardized Workflow for iMSC Generation
The generation of iMSCs from established iPSC lines follows a controlled, multi-stage differentiation process designed to mimic embryonic mesoderm development. Adherence to standardized protocols is critical for ensuring the consistent production of iMSC populations that meet predefined quality specifications.
Critical Quality Control Assays for iMSC Characterization
To ensure iMSCs meet the necessary standards for therapeutic application, a battery of quality control assays must be performed. These assays confirm that the cells possess the defining biological properties of MSCs and are safe for clinical use.
1. Immunophenotyping by Flow Cytometry
- Purpose: To verify the expression of canonical MSC surface markers and absence of hematopoietic contaminants.
- Methodology: Cells are harvested, incubated with fluorochrome-conjugated antibodies, and analyzed via flow cytometry.
- Acceptance Criteria: ≥95% positive for CD73, CD90, CD105; ≤2% positive for CD34, CD45, CD11b, CD19, HLA-DR [2].
2. Trilineage Differentiation Assay
- Purpose: To functionally confirm the multipotent differentiation capacity of iMSCs.
- Osteogenic Differentiation: Culture in basal medium supplemented with dexamethasone, ascorbate-2-phosphate, and β-glycerophosphate for 2-3 weeks. Stain with Alizarin Red S to detect calcium deposits [2].
- Chondrogenic Differentiation: Pellet culture in medium with TGF-β3, dexamethasone, ascorbate-2-phosphate, and proline for 3-4 weeks. Stain with Alcian Blue or Toluidine Blue for proteoglycan detection.
- Adipogenic Differentiation: Culture in medium containing dexamethasone, indomethacin, and insulin for 2-3 weeks. Stain with Oil Red O to visualize lipid vacuoles [120].
3. Karyotyping and Genetic Stability Analysis
- Purpose: To ensure the absence of chromosomal abnormalities acquired during iPSC culture or the differentiation process.
- Methodology: G-band karyotyping or higher-resolution techniques like array CGH at various passages post-differentiation.
Research Reagent Solutions for iMSC Standardization
Standardized, high-quality reagents are fundamental to reproducible iMSC research and development. The table below details essential materials and their functions in establishing robust iMSC generation and expansion protocols.
Table 3: Essential Research Reagents for iMSC Work
| Reagent/Category | Specific Examples | Function & Importance |
|---|---|---|
| Cell Culture Media | IMDM, DMEM/F12, StemMACS MSC Expansion Media | Provides essential nutrients and base environment for cell growth [33]. |
| Critical Media Supplements | Human Platelet Lysate (hPL), FGF-2, EGF | hPL replaces FBS, eliminating xenogenic risks and enhancing proliferation. Growth factors maintain MSC phenotype and potency [33]. |
| Differentiation Kits | Osteo-, Chondro-, Adipo-Differentiation Kits (e.g., MilliporeSigma, Thermo Fisher) | Standardized, pre-mixed media components ensure consistent and efficient trilineage differentiation for functional QC [120]. |
| Characterization Antibodies | Anti-CD73, CD90, CD105, CD34, CD45, HLA-DR (e.g., BD Biosciences, BioLegend) | Essential for flow cytometric immunophenotyping to confirm MSC identity per ISCT criteria [2]. |
| Dissociation Reagents | Trypsin/EDTA, TrypLE Select, Accutase | Enzymatic passaging of adherent iMSCs. Animal-origin-free options enhance regulatory compatibility. |
| Bioreactor Systems | Stirred-tank bioreactors, Hollow-fiber systems | Enable scalable, controlled, and automated 3D expansion of iMSCs for clinical-scale manufacturing [121]. |
Industrial Manufacturing and Regulatory Pathway
The transition from laboratory-scale iMSC culture to industrial manufacturing requires a strategic shift from planar culture systems to closed, automated bioreactor platforms. This shift is critical for achieving the volume, consistency, and cost-effectiveness required for commercial viability.
Scalable Manufacturing Process
A standardized industrial manufacturing workflow integrates upstream processing (cell expansion) and downstream processing (harvesting and formulation) into a closed-system pathway to minimize contamination risk and ensure product consistency.
Regulatory Considerations and Standardization
For regulatory approval, manufacturers must demonstrate rigorous control over the entire production process. Key elements include:
- Defined Critical Quality Attributes: Establishing specifications for identity (surface markers), purity (absence of contaminants), viability, and potency (e.g., immunomodulatory activity) [121].
- Process Analytical Technologies: Implementing in-line, at-line, and off-line monitoring to ensure process consistency and final product quality.
- Standardized Potency Assays: Developing quantitative, clinically relevant bioassays that predict the therapeutic efficacy of the final iMSC product, a current challenge in the field [121].
iPSC-derived MSCs represent a transformative advancement for the commercialization of MSC-based therapies, directly addressing the critical issues of scalability, standardization, and quality control that have hampered primary MSC products. The evolving ecosystem of key players, along with the development of robust experimental protocols and scalable manufacturing platforms, is paving the way for a new generation of standardized, off-the-shelf regenerative medicines.
The future trajectory of the field will focus on further enhancing manufacturing efficiency through innovation in bioreactor design and media formulations, establishing universally accepted potency assays, and navigating the regulatory pathway for the first iMSC-based clinical approvals. As these elements converge, iMSCs are poised to fulfill their promise as a reliable, standardized, and powerful tool for researchers and drug development professionals, ultimately enabling more effective and accessible cellular therapies for patients worldwide.
Conclusion
The choice between bone marrow and adipose tissue as an MSC source is not a matter of superiority but of strategic application. Robust comparative data confirms that AD-MSCs typically offer advantages in proliferation yield and ease of harvest, while BM-MSCs often demonstrate superior inherent capacity for osteogenic and chondrogenic differentiation. The field is rapidly evolving beyond simple cell transplantation, with the future pointing towards precision-enhanced MSCs through genetic engineering and preconditioning, as well as the standardized use of cell-derived products like extracellular vesicles. Overcoming the persistent challenges of functional heterogeneity, manufacturing standardization, and the development of predictive potency assays remains the critical path forward. For researchers and clinicians, a nuanced understanding of the distinct biological and functional profiles of BM-MSCs and AD-MSCs is paramount for designing the next generation of effective, reliable MSC-based therapies.
References
- 1. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement - PubMed [https://pubmed.ncbi.nlm.nih.gov/16923606/]
- 2. Mesenchymal stem cells in treating human diseases [https://www.nature.com/articles/s41392-025-02313-9]
- 3. Mesenchymal stem cells: opening a new chapter in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465317/]
- 4. Clarification of the nomenclature for MSC: The International ... [https://pubmed.ncbi.nlm.nih.gov/16236628/]
- 5. Comparative analysis of human Mesenchymal Stromal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616983/]
- 6. Comparison of biological properties of human adipose tissue ... [https://cardiab.biomedcentral.com/articles/10.1186/s12933-025-02943-x]
- 7. Characterisation of mesenchymal stromal cells in clinical trial reports: analysis of published descriptors | Stem Cell Research & Therapy [https://link.springer.com/article/10.1186/s13287-021-02435-1]
- 8. Defining minimal criteria for peer-reviewed reporting of mesenchymal stromal cell clinical trials for autoimmune diseases - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S1465324925007443]
- 9. Validated methods for isolation and qualification of ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06972-8]
- 10. Comparative characteristic study from bone marrow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8636658/]
- 11. Mesenchymal Stromal Cells: Markers, Isolation and Culture ... [https://www.stemcell.com/mesenchymal-cells-lp.html]
- 12. Isolation, differentiation, and characterization of mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5660271/]
- 13. Expansion and characterization of bone marrow derived ... [https://www.nature.com/articles/s41598-021-83088-1]
- 14. and Adipose Tissue-Derived Mesenchymal Stem Cells under ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0334482]
- 15. Comparison of biological properties of human adipose ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574098/]
- 16. Bone marrow vs. adipose MSCs - Research Article [https://www.foreverlabs.com/articles/blog/bone-marrow-vs-adipose-mscs]
- 17. Adipose derived stem cells – Sources, differentiation ... [https://www.sciencedirect.com/science/article/pii/S0753332225002306]
- 18. Adipose-derived stem cell exosomes: emerging roles and ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1637342/full]
- 19. Therapeutic and Clinical Potential of Adipose-Derived ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12610433/]
- 20. Adipose-derived and bone marrow mesenchymal stem cells [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-018-0914-1]
- 21. Adipose-derived mesenchymal stem cells (MSCs) are a ... [https://www.sciencedirect.com/science/article/pii/S2452199X23003997]
- 22. Adipose and Bone Marrow Derived-Mesenchymal Stromal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9902123/]
- 23. Adipose-derived mesenchymal stem cells and their ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1617157/full]
- 24. Pivotal Trends Impacting the Global Cord Blood Industry in ... [https://bioinformant.com/cord-blood-trends/]
- 25. Menstrual blood and endometrial mesenchymal stem ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925004807]
- 26. Research on the mechanism of human umbilical cord ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04773-w]
- 27. Menstrual blood-derived mesenchymal stromal cell ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1643408/full]
- 28. Endometrial regeneration using menstrual blood-derived ... [https://www.sciencedirect.com/science/article/pii/S2352320425000732]
- 29. Adipose Tissue and Bone Marrow-Derived Mesenchymal ... [https://pubmed.ncbi.nlm.nih.gov/38441812/]
- 30. Stem Cell Therapy: Overview, Benefits & Risks (2025) [https://www.dvcstem.com/post/stem-cell-therapy]
- 31. Mesenchymal stem cells: paracrine signaling and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2902653/]
- 32. Mesenchymal stem cell [https://en.wikipedia.org/wiki/Mesenchymal_stem_cell]
- 33. Comparative analysis of human mesenchymal stem cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4453294/]
- 34. Trilineage Differentiation of Multipotent Human ... [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/cell-culture-and-cell-culture-analysis/primary-cell-culture/mesenchymal-stem-cell-differentiation?srsltid=AfmBOooZvPSUyHyTX36bK0U7UcgqEiibBwepa3R1tyuXm-Cvhtsgdpo1]
- 35. Effects of microenvironment and biological behavior on the ... [https://www.sciencedirect.com/science/article/pii/S235230422300140X]
- 36. Frontiers | Immunomodulation by Mesenchymal Stem Cells... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.01191/full]
- 37. Emerging Therapeutic Approaches For Multipotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3748957/]
- 38. by Mesenchymal Stem Cells... Immunomodulation [https://pubmed.ncbi.nlm.nih.gov/31214172/]
- 39. Mesenchymal Stem Cells Derived from Adipose Tissue vs ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0058198]
- 40. Clinical Protocols for the Isolation and Expansion of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3076361/]
- 41. An improved protocol for isolation and culture of mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5982388/]
- 42. Translation of a standardized manufacturing protocol for ... [https://www.sciencedirect.com/science/article/pii/S1465324919300374]
- 43. Towards the standardization of human platelet lysate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12245862/]
- 44. Human Platelet Lysate as Alternative of Fetal Bovine ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.915277/full]
- 45. Human platelet lysate as an alternative to fetal bovine ... [https://pubmed.ncbi.nlm.nih.gov/31059024/]
- 46. Lyophilized human platelet lysate: manufacturing, quality ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1513444/full]
- 47. Human platelet lysate standardization across three ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04445-9]
- 48. Evaluation of platelet lysate as a substitute for FBS in ... [https://www.nature.com/articles/s41598-018-30772-4]
- 49. Human platelet lysate combined with mesenchymal stem ... [https://www.nature.com/articles/s41598-024-79050-6]
- 50. Molecular mechanisms of mesenchymal stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3812518/]
- 51. Adipogenesis, Osteogenesis, and Chondrogenesis of Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7362937/]
- 52. MGP regulates the adipogenic differentiation of ... [https://www.nature.com/articles/s41420-025-02472-2]
- 53. How mesenchymal stem cells transform into adipocytes [https://pmc.ncbi.nlm.nih.gov/articles/PMC10989283/]
- 54. Osteogenic differentiation of human mesenchymal stromal ... [https://www.nature.com/articles/s41598-021-91501-y]
- 55. Chondrogenic Differentiation of Mesenchymal Stem Cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC4241862/]
- 56. Chondrogenic differentiation of mesenchymal stem cells ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.991855/full]
- 57. A comparison of adult and neonatal tissue-derived MSC | Cell ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-9-12]
- 58. Detailed characterization of bone marrow adipose tissue ... [https://pubmed.ncbi.nlm.nih.gov/39894290/]
- 59. Mesenchymal stem/stromal cells as next-generation drug ... delivery [https://pubmed.ncbi.nlm.nih.gov/34311638/]
- 60. Human amniotic mesenchymal stromal cells (hAMSCs) as potential... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-015-0140-z]
- 61. Mesenchymal Stem Cells Isolated from Adipose and Other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3361347/]
- 62. Emerging Strategies for Cargo Loading and Engineering of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472437/]
- 63. Mesenchymal stromal cell extracellular vesicles as immune ... [https://www.sciencedirect.com/science/article/pii/S0166223625001948]
- 64. Enhancing extracellular vesicle cargo loading and ... [https://www.nature.com/articles/s41467-024-49678-z]
- 65. Mesenchymal Stem Cell Therapy and Its Clinical ... [https://www.intechopen.com/online-first/1228919]
- 66. Engineering strategies for customizing extracellular vesicle ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-022-02806-2]
- 67. A review on mesenchymal stem cells and their secretome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12234943/]
- 68. From bench to bedside: translating mesenchymal stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12344367/]
- 69. Mesenchymal Stem Cells Market Size, Share & Trends 2035 [https://www.researchnester.com/reports/mesenchymal-stem-cells-market/3933]
- 70. 4 Breakthrough Late-stage GvHD Drugs in Development [https://www.delveinsight.com/blog/gvhd-drugs-in-development]
- 71. Obesity‐Induced Loss of Function of Bone Marrow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12331444/]
- 72. Mesenchymal Stem Cells and Their Derivatives [https://www.mdpi.com/2673-8449/5/4/37]
- 73. Functionalized mesenchymal stem cells for enhanced bone ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04699-3]
- 74. A scalable platform for EPSC-Induced MSC extracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12326622/]
- 75. Understanding the molecular basis of mesenchymal stem ... [https://www.nature.com/articles/s41419-025-08094-x]
- 76. Overcoming challenges in MSC-sEV therapeutics [https://www.sciencedirect.com/science/article/pii/S1465324925005912]
- 77. Research Insights - University of Georgia Office of Research [https://research.uga.edu/research-insights/collaborative-research-investigating-the-role-and-interplay-of-microenvironment-manufacturing-and-metabolism-on-msc-production-of-extracellular-vesicles/]
- 78. iPSC-, MSC- and iMSC-Derived Exosome Therapeutics [https://www.reprocell.com/blog/ipsc-msc-and-imsc-derived-exosome-therapeutics-the-cell-free-future-of-regenerative-medicine]
- 79. Hypoxia and inflammatory factor preconditioning enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10696190/]
- 80. Priming approaches to improve the efficacy of ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-019-1224-y]
- 81. First Clinical Experiences Using Preconditioning Approaches to Improve MSC-Based Therapies | Current Stem Cell Reports [https://link.springer.com/article/10.1007/s40778-023-00232-5]
- 82. Preconditioning of Mesenchymal Stem Cells Enhances the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11504366/]
- 83. Inflammatory priming of mesenchymal stromal cells ... [https://www.sciencedirect.com/science/article/pii/S0006291X25011064]
- 84. Dexamethasone priming enhances stemness and immunomodulatory property of tissue-specific human mesenchymal stem cells - PubMed [https://pubmed.ncbi.nlm.nih.gov/34736395/]
- 85. Enhancing mesenchymal stem cell survival and homing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10478247/]
- 86. Mesenchymal Stromal Cell Homing: Mechanisms and ... [https://www.sciencedirect.com/science/article/pii/S2589004219301415]
- 87. Mesenchymal stem cells from human bone marrow or adipose tissue differently modulate mitogen-stimulated B-cell immunoglobulin production in vitro - PubMed [https://pubmed.ncbi.nlm.nih.gov/18262807/]
- 88. The secretion profile of mesenchymal stem cells and ... [https://www.nature.com/articles/s41392-022-00932-0]
- 89. Trends in mesenchymal stem cell-derived extracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12488731/]
- 90. Extracellular vesicle-based drug overview [https://www.nature.com/articles/s41392-025-02312-w]
- 91. Engineered mesenchymal stem cell-derived exosomes [https://www.sciencedirect.com/science/article/pii/S2405580825004005]
- 92. Mesenchymal stem cells biological and biotechnological ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299924004072]
- 93. Quantifiable Metrics for Predicting MSC Therapeutic Efficacy [https://pmc.ncbi.nlm.nih.gov/articles/PMC5260823/]
- 94. Attitudes Towards Standardization of Mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10630078/]
- 95. Challenges in Clinical Development of Mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6811694/]
- 96. Challenges and advances in clinical applications of ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-021-01037-x]
- 97. Current applications of adipose-derived mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9490047/]
- 98. Potency assay to predict the anti-inflammatory capacity of a ... [https://www.sciencedirect.com/science/article/pii/S1465324924000501]
- 99. Comparing the Immunomodulatory Properties of Bone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4630659/]
- 100. Current progress and limitations of research regarding the ... [https://link.springer.com/article/10.1007/s43994-024-00147-9]
- 101. Lineage Differentiation Potential of Different Sources ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9028477/]
- 102. Tissue source determines the differentiation potentials of ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-017-0716-x]
- 103. New Insights into Osteogenic and Chondrogenic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3676919/]
- 104. Respective stemness and chondrogenic potential of ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-020-01786-5]
- 105. A Comparative Study of the Effect of Anatomical Site on ... [https://www.mdpi.com/2073-4409/10/9/2469]
- 106. Comparison of Osteogenic and Chondrogenic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5960732/]
- 107. Mesenchymal stem cell secretome for regenerative medicine [https://www.sciencedirect.com/science/article/pii/S2090123224001814]
- 108. Exploring the therapeutic potential of MSC-derived ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04616-8]
- 109. Immunomodulatory and Regenerative Functions of MSC ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383523/]
- 110. Unlocking the Therapeutic Potential of Adipose-Derived ... [https://www.mdpi.com/2079-7737/13/12/1016]
- 111. Human bone marrow- and adipose-mesenchymal stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4529699/]
- 112. The Immunomodulatory mechanism and research progress of ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04333-2]
- 113. Immunomodulatory Functions of Mesenchymal Stem Cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12572674/]
- 114. A systematic review of mesenchymal stem cell secretome [https://www.sciencedirect.com/science/article/pii/S8756328224002588]
- 115. Adipose-derived mesenchymal stromal cells in clinical trials [https://www.sciencedirect.com/science/article/pii/S0024320524003515]
- 116. Mesenchymal Stem Cells for Regenerative Medicine - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6721852/]
- 117. Different Sources of Mesenchymal Stem Cells for Tissue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9181542/]
- 118. Trends in mesenchymal stem cell-derived extracellular ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1625787/full]
- 119. Krüppel-like factor 4 regulates cellular proliferation and ... [https://www.sciencedirect.com/science/article/pii/S2405580825003280]
- 120. Induced Mesenchymal Stem Cells: An Emerging Source for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11943107/]
- 121. Mesenchymal Stem Cells/Medicinal Signaling ... [https://www.businesswire.com/news/home/20251009529610/en/Mesenchymal-Stem-CellsMedicinal-Signaling-Cells-Advances-Applications-Report-2025-1670-Trials-12-Approvals-Worldwide-and-Path-to-50-Approvals-by-2040---ResearchAndMarkets.com]