hiPSCs in Cardiovascular Disease Modeling: From Patient-Specific Cells to Precision Drug Discovery
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) are revolutionizing cardiovascular research by providing a patient-specific, human-relevant platform for disease modeling and drug development.
hiPSCs in Cardiovascular Disease Modeling: From Patient-Specific Cells to Precision Drug Discovery
Abstract
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) are revolutionizing cardiovascular research by providing a patient-specific, human-relevant platform for disease modeling and drug development. This article explores the foundational principles of hiPSC technology, detailing the reprogramming of somatic cells and their differentiation into cardiomyocytes. It examines advanced methodological applications in modeling inherited arrhythmias, cardiomyopathies, and drug-induced cardiotoxicity, while also addressing key challenges such as cellular immaturity and phenotypic variability. Furthermore, it validates hiPSC-CMs against traditional models, highlighting their superior predictive value for human physiology. Aimed at researchers, scientists, and drug development professionals, this review synthesizes current advancements and future directions, underscoring the role of hiPSCs in advancing personalized medicine and improving the efficacy and safety of cardiovascular therapeutics.
The Foundation of iPSC Technology: Reprogramming and Cardiac Differentiation
The discovery that somatic cells can be reprogrammed to a pluripotent state represents a paradigm shift in regenerative medicine and biomedical research. In 2006, Shinya Yamanaka and his team demonstrated that the forced expression of four specific transcription factors—Oct4, Sox2, Klf4, and c-Myc (collectively known as the Yamanaka factors)—could convert specialized adult cells into induced pluripotent stem cells (iPSCs) [1]. This groundbreaking work, which earned Yamanaka the Nobel Prize in 2012, demonstrated that cellular differentiation is not a unidirectional process and that epigenetic fate can be reversed through specific molecular interventions [2].
In the context of cardiovascular disease research, iPSC technology offers unprecedented opportunities. Scientists can now generate patient-specific cardiomyocytes in vitro, enabling the study of disease mechanisms, cardiotoxicity testing, and the development of personalized therapeutic approaches [3]. This technical guide explores the core principles of somatic cell reprogramming using Yamanaka factors, with specific emphasis on methodologies and applications relevant to cardiovascular disease modeling.
Molecular Mechanisms of the Yamanaka Factors
Core Pluripotency Network
The Yamanaka factors function as master regulators of pluripotency by activating a self-reinforcing transcriptional network that silences somatic cell identity genes while activating embryonic stem cell programs. These four transcription factors work in concert to reshape the epigenetic landscape and initiate the reprogramming cascade [4].
Table 1: Core Yamanaka Factors and Their Functions in Reprogramming
| Transcription Factor | Primary Function in Reprogramming | Role in Pluripotency Maintenance | Associated Risks |
|---|---|---|---|
| Oct4 (Pou5f1) | Establishes and maintains pluripotency; essential for inner cell mass development | Forms core regulatory circuit with Sox2 and Nanog; regulates pluripotency-associated genes | Over-expression causes differentiation; depletion blocks reprogramming |
| Sox2 | Partners with Oct4 to regulate key pluripotency genes | Maintains self-renewal; cooperates with Oct4 in transcriptional activation | Expressed in neural stem cells; implicated in some cancers |
| Klf4 | Facilitates epigenetic remodeling; activates Nanog expression | Enhances core pluripotency factors for developmental regulation; contains zinc-finger DNA-binding domains | Can function as both oncogene and tumor suppressor depending on context |
| c-Myc | Promotes chromatin accessibility; regulates metabolic switching | Orchestrates proliferation and metabolism; distinct role from other factors | Potent oncogene; increases tumorigenesis risk in iPSC-derived tissues |
Mechanism of Action
The reprogramming process initiates when the Yamanaka factors are introduced into somatic cells. c-Myc functions as a pioneer factor that opens chromatin structure by binding to methylated regions, making previously inaccessible genetic sequences available for transcriptional activation [4]. Subsequently, Oct4 and Sox2 interact with enhancers and promoters of genes involved in somatic cell identity and pluripotency, forming heterodimers that recognize composite SOX-OCT binding sites in the genome [5] [4].
This transcription factor partnership creates a self-reinforcing interconnected loop that activates their own promoters along with those of other pluripotency-associated genes such as Nanog [4]. During this process, mesenchymal genes are silenced while epithelial genes such as Cdh1, Epcam, and Ocln are upregulated, facilitating the mesenchymal-to-epithelial transition that characterizes early reprogramming stages [4]. The Yamanaka factors collectively regulate a developmental signaling network composed of at least 16 crucial developmental signaling pathways to maintain pluripotency, including previously unknown pathways in ES cells such as apoptosis and cell-cycle regulation [5].
Figure 1: Molecular Reprogramming Cascade by Yamanaka Factors
Reprogramming Methodologies and Protocols
Delivery Systems for Reprogramming Factors
Multiple delivery systems have been developed to introduce the Yamanaka factors into somatic cells, each with distinct advantages and limitations for research and clinical applications.
Table 2: Comparison of Yamanaka Factor Delivery Methods
| Delivery Method | Integration into Genome | Reprogramming Efficiency | Safety Profile | Best Use Cases |
|---|---|---|---|---|
| Retroviral/Lentiviral | Yes (Random integration) | High | Low (Oncogenic risk) | Basic research, difficult-to-reprogram cells |
| Sendai Virus | No (Non-integrating RNA virus) | High | Moderate (Viral persistence concerns) | Clinical applications, disease modeling |
| Episomal Vectors | No (Extrachromosomal) | Low to moderate | High | Clinical applications, GMP compliance |
| Synthetic mRNA | No (Transient expression) | High with optimization | High | Clinical applications, personalized medicine |
| Small Molecules | No (Chemical induction) | Low to moderate | High (But specificity challenges) | Research, safety-sensitive applications |
Detailed Reprogramming Protocol Using mRNA Technology
The synthetic mRNA reprogramming method represents one of the safest and most efficient approaches for generating clinical-grade iPSCs, particularly for cardiovascular applications where genetic stability is paramount [6] [7].
Protocol: mRNA-Based Reprogramming of Human Fibroblasts
Day 0: Target Cell Seeding
- Plate human dermal fibroblasts or cardiac fibroblasts in 6-well plates at optimal seeding density (1×10^5 to 1×10^6 cells per well) to achieve 60-80% confluency after 24 hours [7].
Day 1: B18R Protein Pretreatment and mRNA Transfection
- Pretreat cells with 200 ng/mL B18R protein in DMEM for 2 hours to inhibit innate immune responses against introduced RNA [7].
- Prepare transfection complex: Combine 0.5 μL VEE-OKS-iG RNA (encoding OCT4, KLF4, SOX2), 0.5 μL B18R RNA, and 4.0 μL RiboJuice mRNA transfection reagent in 250 μL Opti-MEM medium [7].
- Add RNA-transfection reagent complexes dropwise to cells and incubate for 4 hours at 37°C, 5% CO₂ [7].
- Replace with DMEM supplemented with 10% FBS, 1X glutamine, and 200 ng/mL B18R protein [7].
Days 2-11: Puromycin Selection and Daily Transfections
- Replace medium daily with fresh DMEM containing 200 ng/mL B18R protein and puromycin at the optimal selection concentration (determined empirically for each cell line) [7].
- Monitor cell death and morphology changes; by days 4-5, approximately 30-60% cell death should be observable [7].
- Continue daily transfections and puromycin selection until day 11 [7].
Days 11-18: Replating and Expansion
- When puromycin-resistant cells reach 70-90% confluency (typically between days 9-18), replate them onto Matrigel-coated plates or inactivated MEF feeders [7].
- Culture in MEF-conditioned medium supplemented with 10 ng/mL bFGF, 1X human iPSC reprogramming enhancer, and 200 ng/mL B18R protein [7].
Days 18-30: iPSC Colony Picking and Expansion
- Monitor for emergence of compact colonies with defined borders, typically appearing between days 15-20 [7].
- Pick individual colonies when they reach approximately 200 cells and transfer to PluriSTEM Human ES/iPSC medium for further expansion and characterization [7].
Figure 2: mRNA Reprogramming Workflow Timeline
Enhanced Protocol for Senescent and Pathologic Cells
Reprogramming efficiency decreases significantly when working with senescent or pathologic cells, which is particularly relevant for cardiovascular disease modeling where patient-specific cells often come from elderly individuals or those with chronic conditions [8]. An enhanced protocol has been developed specifically for such challenging cases:
Enhanced Protocol for Senescent Cardiac Fibroblasts
- Use protocol modifications including culture medium supplemented with SB431542 (a TGF-β inhibitor) instead of TGF-β [8].
- Employ an expanded factor combination including OCT3/4, short hairpin p53, SOX2, KLF4, LIN28, L-MYC, Glis-1, and Tet-1 [8].
- This approach successfully generated iPSCs from myocardial fibroblasts isolated from all four chambers of a pathologic heart from a heart transplant recipient with dilated cardiomyopathy, whereas conventional protocols failed [8].
Applications in Cardiovascular Disease Modeling
iPSC-Derived Cardiomyocytes for Disease Modeling
The generation of human iPSC-derived cardiomyocytes (hiPSC-CMs) has created unprecedented opportunities for cardiovascular disease modeling and drug discovery. Cardiovascular diseases cause approximately 19.8 million deaths annually, ranking as the leading cause of death worldwide, yet the number of new cardiovascular drugs has been steadily decreasing [3]. This decline is partly attributable to the lack of preclinical models that accurately evaluate therapeutic efficacy and safety in humans [3].
hiPSC-CMs enable researchers to reproduce disease-specific characteristics in culture dishes, offering several advantages over traditional models:
- Species-specific relevance: Animal models have limited predictive value due to differences in cardiac biology between species (e.g., mouse heart rate is approximately eight times higher than humans, with different ion current dependencies) [3].
- Patient-specific modeling: hiPSCs can be generated from patients with specific genetic cardiovascular disorders, creating personalized disease models [3].
- Unlimited expansion potential: Unlike primary human cardiomyocytes that dedifferentiate in culture, hiPSCs offer a renewable cell source [3].
Current Limitations and Maturation Challenges
Despite their promise, hiPSC-CMs exhibit an immature phenotype similar to fetal cardiomyocytes, which limits their application in disease modeling and drug discovery [3]. Key differences between hiPSC-CMs and adult human cardiomyocytes include:
Table 3: Immaturity of hiPSC-Derived Cardiomyocytes and Functional Consequences
| Characteristic | hiPSC-Derived Cardiomyocytes | Adult Human Cardiomyocytes | Functional Impact |
|---|---|---|---|
| Cell Morphology | Small (3000-6000 μm³), rounded shape | Cylindrical (∼40,000 μm³) | Reduced contractile force generation |
| Sarcomere Organization | Poorly organized, randomly oriented | Form myofibrils parallel to entire cell | Impaired coordinated contraction |
| Sarcomere Isoforms | Express fetal isoforms (αMHC, MLC2a, ssTnI) | Express adult isoforms (βMHC, MLC2v, cTnI) | Altered contractile kinetics and calcium sensitivity |
| T-Tubules | Rarely observed | Well-developed regular T-tubule network | Delayed calcium-induced calcium release (CICR) |
| Metabolism | Primarily glycolytic | Primarily oxidative phosphorylation | Different energy utilization and stress responses |
| Electrophysiology | Fetal-like ion channel expression | Adult-specific ion channel patterns | Different repolarization properties and drug responses |
Maturation Strategies for Enhanced Disease Modeling
Multiple strategies have been developed to enhance the maturation of hiPSC-CMs to better model adult cardiovascular diseases:
- Long-term culture: Extended culture duration (up to 120 days) promotes structural and functional maturation [3].
- Biochemical stimulation: Treatment with thyroid hormone (T3), glucocorticoids, and cAMP inducers promotes adult-like gene expression patterns [3].
- Biophysical stimulation: Application of mechanical stretch, electrical pacing, and substrate stiffness tuning enhances structural alignment and maturation [3].
- 3D tissue engineering: Creating engineered heart tissues using biomaterial scaffolds and multicellular compositions improves tissue-level organization and function [3].
- Metabolic manipulation: Shifting culture conditions to promote fatty acid oxidation instead of glycolysis drives metabolic maturation [3].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for Yamanaka Factor Reprogramming
| Reagent Category | Specific Examples | Function in Reprogramming | Application Notes |
|---|---|---|---|
| Reprogramming Factors | VEE-OKS-iG RNA (OCT4, KLF4, SOX2); c-Myc expression vector | Core transcription factors inducing pluripotency | Optimal ratio crucial for efficiency; c-Myc can be omitted but reduces efficiency |
| Immune Suppressors | B18R protein | Inhibits innate immune response against introduced RNA | Critical for mRNA-based methods; improves cell survival during transfection |
| Delivery Reagents | RiboJuice mRNA transfection reagent; Lipofectamine STEM | Facilitates cellular uptake of reprogramming factors | Optimization required for different cell types; can impact cytotoxicity |
| Selection Agents | Puromycin; Geneticin (G418) | Selects for successfully transfected cells | Concentration must be empirically determined for each cell type |
| Culture Media | PluriSTEM Human ES/iPSC medium; MEF-conditioned medium | Supports pluripotent stem cell growth and maintenance | Often requires supplementation with bFGF for optimal results |
| Enhanced Factors | LIN28, L-MYC, Glis-1, Tet-1 | Improves reprogramming efficiency in difficult cells | Particularly valuable for senescent or pathologic somatic cells |
| Signaling Modulators | SB431542 (TGF-β inhibitor); Ascorbic acid | Enhances reprogramming efficiency; reduces senescence | Context-dependent effects; optimal concentration varies by cell type |
Reprogramming somatic cells using Yamanaka factors has revolutionized cardiovascular research by enabling the generation of patient-specific iPSCs that can be differentiated into cardiomyocytes for disease modeling and drug screening. The continued refinement of reprogramming methodologies—particularly the development of non-integrating delivery systems and enhanced protocols for senescent cells—has improved the safety and efficiency of iPSC generation.
Future directions in this field include the development of more precise temporal control over factor expression, further reduction in genomic instability risks, and the creation of increasingly mature cardiomyocyte models that better recapitulate adult heart physiology. As these technologies converge with advances in gene editing, tissue engineering, and machine learning, iPSC-based cardiovascular disease modeling is poised to become an indispensable tool for drug discovery and personalized medicine, potentially transforming how we understand and treat the world's leading cause of mortality.
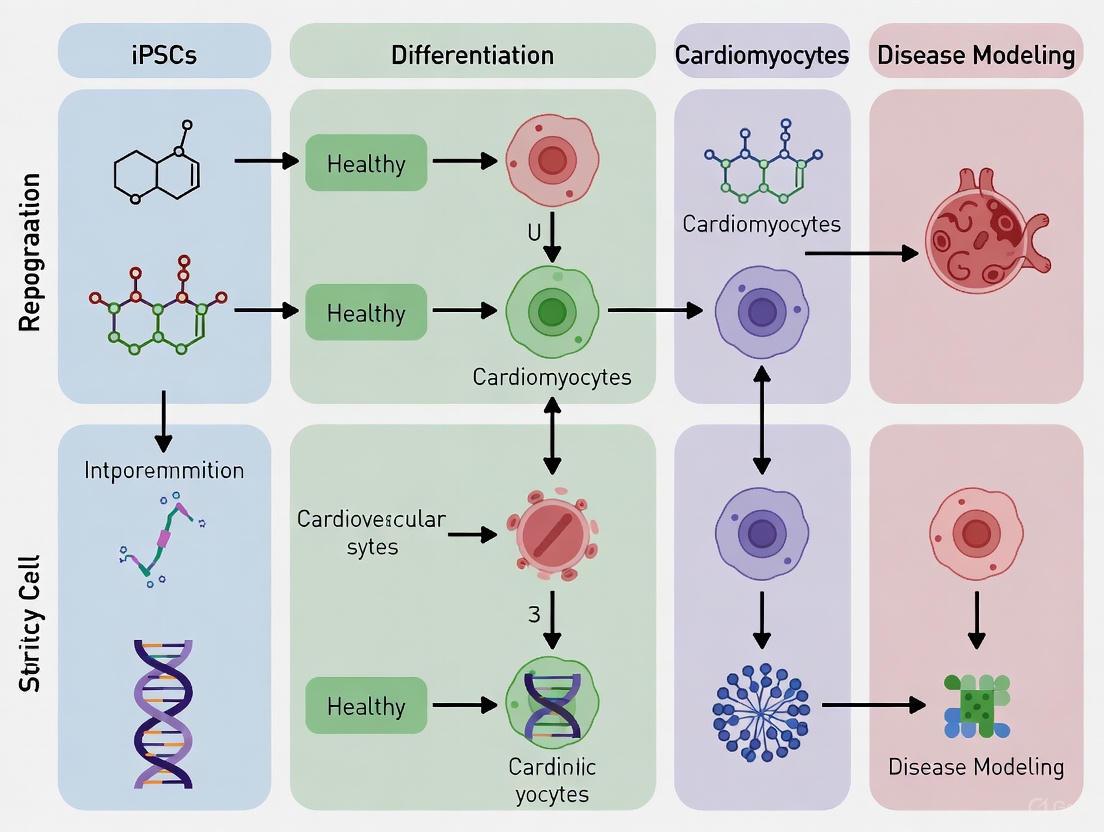
The selection of an appropriate somatic cell source is a critical first step in the successful generation of human induced pluripotent stem cells (iPSCs) for cardiovascular disease modeling. This initial decision directly influences reprogramming efficiency, the quality of the resulting iPSC lines, and their subsequent applicability in downstream research and potential therapies [9]. The ability to derive patient-specific cardiomyocytes from iPSCs has provided an unprecedented platform for modeling inherited cardiac disorders, screening pharmacological compounds, and developing personalized therapeutic strategies [10]. Since the groundbreaking discovery of iPSC technology by Shinya Yamanaka in 2006, which demonstrated that somatic cells could be reprogrammed into a pluripotent state using defined factors, the field has rapidly diversified the types of somatic cells used for reprogramming [11] [12]. This technical guide provides an in-depth comparison of three prominent somatic cell sources—dermal fibroblasts, peripheral blood mononuclear cells, and urine-derived renal epithelial cells—within the specific context of cardiovascular research applications. We evaluate these sources based on quantitative metrics, detail optimized experimental protocols, and discuss their relative advantages and limitations for generating iPSC-derived cardiovascular cells.
The ideal somatic cell source for iPSC generation balances reprogramming efficiency, patient convenience, scalability, and genomic stability. The table below provides a systematic comparison of the three primary cell sources evaluated in this guide.
Table 1: Comparative Analysis of Somatic Cell Sources for iPSC Generation
| Parameter | Dermal Fibroblasts | Peripheral Blood Mononuclear Cells (PBMCs) | Urine-Derived Renal Epithelial Cells |
|---|---|---|---|
| Reprogramming Efficiency | Moderate | Comparable to fibroblasts [9] | Robust [9] |
| Invasiveness of Collection | Invasive (skin biopsy) [9] | Minimally invasive (blood draw) [9] | Non-invasive (urine sample) [9] |
| Cell Yield | Readily expanded [9] | Varies by donor | Sufficient for multiple reprogramming cycles [9] |
| Genomic Stability | High [9] | High | Not Specified |
| Reprogramming Factors | OSKM (Oct4, Sox2, Klf4, c-Myc) [9] | OSKM [9] | OSKM [9] |
| Key Advantages | High genomic stability, reliable protocol [9] | Minimally invasive, established immune cell protocols [9] | Completely non-invasive, easily repeatable sampling [9] |
| Primary Limitations | Invasive collection, requires tissue culture expansion [9] | Lower yield for some blood cell types, requires specific mobilization in some cases | Lower initial cell number, requires optimized culture conditions |
Detailed Methodologies for Cell Isolation and Reprogramming
Somatic Cell Isolation and Culture
Dermal Fibroblasts: Fibroblasts are typically obtained via a 3-4 mm punch biopsy from the dermis. The tissue explant is minced and cultured in fibroblast medium (e.g., Dulbecco's Modified Eagle Medium supplemented with 10-15% fetal bovine serum, 2 mM L-glutamine, and 1% penicillin/streptomycin) [9]. Explants are maintained at 37°C with 5% CO₂, and outgrowing fibroblasts are passaged upon reaching 70-80% confluence using trypsin-EDTA. Early-passage fibroblasts (passages 3-5) are optimal for reprogramming to minimize the accumulation of epigenetic alterations [9].
Peripheral Blood Mononuclear Cells (PBMCs): Blood samples are collected in anticoagulant-treated tubes (e.g., EDTA or heparin). PBMCs are isolated via density-gradient centrifugation using Ficoll-Paque. For T lymphocyte reprogramming, cells can be stimulated with cytokines like IL-7 and stem cell factor [9]. Alternatively, erythroblasts expanded in specific cytokine cocktails (e.g., SCF, EPO, IL-3, IL-6, and dexamethasone) can serve as an efficient starting population [9].
Urine-Derived Renal Epithelial Cells: Mid-stream urine samples (typically 100-200 mL) are collected in sterile containers. Cells are harvested by centrifugation and initially cultured in specialized media such as REGM (Renal Epithelial Cell Growth Medium) or a defined cocktail of keratinocyte serum-free medium (KSFM) and fibroblast medium [9]. The initial emergence of epithelial-like colonies usually occurs within 5-10 days, after which they can be expanded for reprogramming.
Reprogramming to Pluripotency
The core reprogramming process involves the introduction of specific transcription factors to revert somatic cells to a pluripotent state. The original Yamanaka factors (OSKM: Oct4, Sox2, Klf4, c-Myc) remain a standard combination [9] [13] [12]. Multiple delivery systems have been developed, each with distinct advantages for safety and efficiency, as summarized in the table below.
Table 2: Delivery Systems for Reprogramming Factors
| Vector/Platform | Genetic Material | Genomic Integration | Key Features |
|---|---|---|---|
| Retrovirus | RNA | Yes | High efficiency, but silencing required; risk of insertional mutagenesis [9] [13]. |
| Lentivirus | RNA | Yes | Can infect non-dividing cells; risk of insertional mutagenesis [9]. |
| Sendai Virus | RNA | No | High efficiency, non-integrating; diluted out over passaging [9] [13]. |
| Episomal Plasmid | DNA | No | Integration-free; low efficiency, requires repeated transfection [9] [13]. |
| Synthetic mRNA | RNA | No | Non-integrating, high efficiency; can trigger immune responses [9] [13]. |
| Recombinant Protein | Protein | No | Highest safety profile; very low efficiency and technically challenging [9] [13]. |
The reprogramming workflow follows a characteristic sequence, beginning with the silencing of somatic cell genes, followed by the activation of pluripotency networks, and culminating in the emergence of stable iPSC colonies [12].
Diagram 1: The sequential process of somatic cell reprogramming to iPSCs.
Following reprogramming, iPSCs require rigorous quality control before differentiation. This includes verification of pluripotency marker expression (e.g., Oct4, Nanog via PCR or immunostaining) and functional assessment of differentiation potential into the three germ layers [9]. Genomic integrity must be confirmed through karyotyping and other analyses to ensure the cells are free of reprogramming-induced mutations [9].
Cardiac Differentiation and Maturation from iPSCs
Directed Cardiac Differentiation
The most common method for differentiating iPSCs into cardiomyocytes (iPSC-CMs) is via modulation of the Wnt/β-catenin signaling pathway [14]. This highly efficient protocol can be performed in both monolayer and suspension culture formats.
Diagram 2: Key stages of cardiac differentiation via Wnt pathway modulation.
Bioreactor Suspension Differentiation Protocol: Recent advances have optimized cardiac differentiation in stirred suspension bioreactors, which offer superior scalability, reproducibility, and maturation compared to traditional monolayer cultures [14]. The optimized workflow is as follows:
- Input Cell Quality Control: Use quality-controlled master cell banks of iPSCs. High pluripotency marker SSEA4 (>70% by FACS) is correlated with successful differentiation [14].
- Embryoid Body (EB) Formation: Culture hiPSCs in suspension to form EBs. Monitor EB diameter, targeting ~100 µm at the time of Wnt activation for optimal efficiency [14].
- Cardiac Differentiation: Initiate mesoderm differentiation by adding 7 µM CHIR99021 (a GSK-3β inhibitor) for 24 hours. After a 24-hour gap, add 5 µM IWR-1 (a Wnt inhibitor) for 48 hours to specify cardiac mesoderm [14].
- Maintenance and Harvesting: Continue culture in RPMI + B27 supplement (with insulin). Spontaneous contractions typically appear by differentiation day 5 [14].
- Cryopreservation: Cells can be cryopreserved at day 15 post-differentiation with high viability (>90%) upon thawing [14].
This protocol consistently yields ~1.2 million cells per mL with high purity (~94% Troponin T-positive cells) and predominantly ventricular identity [14].
Enhancing Cardiomyocyte Maturity
iPSC-CMs are typically immature, resembling fetal rather than adult cardiomyocytes. Enhancing maturity is crucial for accurate disease modeling. Key strategies include:
- Advanced 3D Culture: Engineering bioactive, anisotropic extracellular matrix (ECM) scaffolds derived from human iPSC-cardiac fibroblasts (hiPSC-CFs) provides a physiologically relevant microenvironment that promotes structural and functional maturation of iPSC-CMs, leading to improved contractile properties and electrophysiology [15].
- Co-culture Systems: Co-culturing iPSC-CMs with hiPSC-CFs in engineered platforms significantly enhances CM contractile strain, organization, and contraction kinetics. This enhancement is mediated by both paracrine signaling and direct cell-cell contact [16].
- Metabolic Maturation: Shifting the culture medium to lactate-based metabolism enriches for cardiomyocytes and promotes a more adult-like metabolic phenotype [14].
- Chronic Stimulation: Applying mechanical stretch [15] and electrical pacing [16] to iPSC-CMs can drive further structural and functional maturation.
The Scientist's Toolkit: Essential Reagents for iPSC-CV Research
Table 3: Key Research Reagent Solutions for iPSC and Cardiac Differentiation Workflows
| Reagent/Category | Example Products | Primary Function |
|---|---|---|
| Reprogramming Vectors | CytoTune-iPS Sendai Virus Reprogramming Kit, Episomal plasmids | Safe and efficient delivery of OSKM reprogramming factors. |
| Cell Culture Media | mTeSR1, StemFlex, Essential 8 (E8) | Maintenance of pluripotent stem cells in defined, feeder-free conditions. |
| Extracellular Matrices | Matrigel, Recombinant Laminin-521 | Coating substrate for adherent culture of iPSCs. |
| Cardiac Differentiation Kits | Gibco PSC Cardiomyocyte Differentiation Kit, STEMdiff Cardiomyocyte Kit | Directed differentiation of iPSCs to cardiomyocytes using optimized protocols. |
| Small Molecule Inducers | CHIR99021 (Wnt activator), IWP-2/IWR-1 (Wnt inhibitors) | Critical for modulating Wnt signaling to drive cardiac differentiation. |
| Cell Culture Supplements | B-27 Supplement, Insulin | Serum-free supplements for cardiomyocyte maintenance and maturation. |
| Characterization Antibodies | Anti-cTnT (Cardiac Troponin T), Anti-α-Actinin, Anti-MLC2v | Immunostaining to confirm cardiomyocyte identity and subtype (ventricular). |
The choice of somatic cell source for iPSC generation is a fundamental decision that shapes the entire trajectory of cardiovascular disease modeling research. Dermal fibroblasts offer a well-established, genomically stable starting material. Peripheral blood cells provide a less invasive alternative with robust reprogramming efficiency, making them suitable for large-scale donor collections. Urine-derived cells present a completely non-invasive option that is easily repeatable, facilitating longitudinal studies from the same donor. The selection should be guided by the specific requirements of the research project, weighing factors such as invasiveness, scalability, and reprogramming efficiency. When combined with advanced cardiac differentiation protocols—particularly scalable suspension systems—and strategies to enhance cardiomyocyte maturity, iPSCs from any of these sources form a powerful platform for elucidating disease mechanisms, accelerating drug discovery, and advancing personalized cardiovascular medicine.
The advent of induced pluripotent stem cell (iPSC) technology has revolutionized biomedical research, offering unprecedented opportunities for disease modeling, drug discovery, and regenerative medicine. In cardiovascular research, the ability to derive patient-specific cardiomyocytes from iPSCs has enabled the precise in vitro modeling of cardiac diseases and the screening of potential therapeutics [9] [3]. The foundation of this technology lies in reprogramming somatic cells to a pluripotent state by introducing specific transcription factors, predominantly OCT4, SOX2, KLF4, and c-MYC (OSKM) [12]. The choice of delivery method for these reprogramming factors is crucial, as it directly impacts the genomic integrity, functional fidelity, and clinical applicability of the resulting iPSCs. This technical guide comprehensively examines the evolution of iPSC delivery methods, from early integrating vectors to contemporary non-integrating platforms, with specific emphasis on their utility in cardiovascular disease modeling research.
Historical Development and Core Principles
The conceptual foundation for iPSC technology was established through seminal experiments demonstrating that cellular identity is maintained by reversible epigenetic mechanisms. John Gurdon's seminal somatic cell nuclear transfer (SCNT) experiments in 1962 demonstrated that a nucleus from a differentiated somatic cell could support the development of an entire organism when transferred into an enucleated egg [12]. This pivotal finding established that genetic information remains intact during development and that epigenetic regulation is reversible. The direct reprogramming of somatic cells became feasible with the groundbreaking work of Shinya Yamanaka, who identified that retroviral-mediated expression of four transcription factors—Oct4, Sox2, Klf4, and c-Myc—could reprogram mouse fibroblasts into pluripotent stem cells [12]. This discovery unlocked the potential to create patient-specific pluripotent cells without embryonic sources, with Yamanaka receiving the Nobel Prize in 2012 for this transformative achievement.
The reprogramming process involves profound epigenetic remodeling and transcriptional reorganization, reverting somatic cells to a pluripotent state through a partially stochastic process [12]. Successful reprogramming entails silencing somatic genes while activating the endogenous pluripotency network, with the endogenous reactivation of the Oct4 promoter serving as a critical stabilization point for the pluripotent state [9]. The initial methods using integrating viral vectors raised significant safety concerns for clinical applications, driving the development of non-integrating alternatives that minimize the risk of insertional mutagenesis and oncogene reactivation [9] [17].
Comparative Analysis of Delivery Methods
Integrating Delivery Methods
Retroviral and Lentiviral Vectors
Early iPSC generation relied heavily on integrating viral vectors, particularly gamma-retroviruses and lentiviruses, which offer high reprogramming efficiency through stable genomic integration of the transgenes [17]. Retroviral vectors (e.g., pMXs) efficiently infect dividing cells but are prone to incomplete transgene silencing after reprogramming, which can interfere with differentiation [17]. Lentiviral vectors provide the advantage of infecting both dividing and non-dividing cells and can accommodate polycistronic cassettes expressing multiple factors from a single promoter [17].
However, the primary safety concern with these methods is insertional mutagenesis, where random integration disrupts tumor suppressor genes or activates oncogenes, potentially leading to malignant transformation [9] [17]. Additionally, persistent expression or reactivation of reprogramming factors, particularly the oncogene c-MYC, can compromise differentiation and increase tumorigenic risk in derived cells [18] [17]. While excisable systems using Cre-lox technology or piggyBac transposons mitigate these concerns, they still leave genetic scars and require careful sequencing to verify complete removal [17].
Table 1: Comparison of Integrating Delivery Methods
| Method | Mechanism | Efficiency | Advantages | Disadvantages | Cardiovascular Research Utility |
|---|---|---|---|---|---|
| Retroviral Vectors | Genomic integration via reverse transcription | High | High efficiency; stable expression | Only infects dividing cells; insertional mutagenesis; incomplete silencing | Limited due to safety concerns; historical significance |
| Lentiviral Vectors | Genomic integration | High | Infects dividing & non-dividing cells; larger cargo capacity | Insertional mutagenesis; heterogeneous clones | Improved with excisable systems but largely superseded |
| piggyBac Transposon | "Cut-and-paste" transposition | Medium | Excisable system; large cargo capacity | Need to verify excision; potential for genomic alterations | Research tool for generating footprint-free lines |
Non-Integrating and DNA-Free Methods
Sendai Viral Vectors
Sendai virus (SeV) vectors represent a highly efficient non-integrating RNA-based system derived from a murine paramyxovirus [17] [19]. As an RNA replication-competent vector that operates entirely in the cytoplasm, SeV poses minimal risk of genomic integration [19]. These vectors provide robust, transient transgene expression ideal for reprogramming, and can be engineered with temperature-sensitive mutations to facilitate eventual clearance from the cell population [17] [19].
In proof-of-concept studies, SeV vectors have successfully generated transgene-free iPSCs from patients with type 1 and type 2 diabetes, including an 85-year-old individual, demonstrating their applicability to aged and diseased somatic cells [19]. The SeV system induces endogenous pluripotency genes while suppressing senescence pathways, facilitating efficient reprogramming [19]. A minor limitation is the potential difficulty in completely clearing the virus from all cells, though temperature-sensitive variants and prolonged passaging achieve elimination in most cases [17].
mRNA-Based Reprogramming
Synthetic mRNA reprogramming represents the safest approach, completely eliminating risks associated with viral vectors and genomic integration [17]. This method involves repeated transfection of cells with modified mRNAs encoding the reprogramming factors, typically with nucleoside modifications to reduce innate immune recognition and improve stability [17]. The protocol requires daily transfections over approximately two weeks, followed by picking of emerging iPSC colonies [17].
While this method offers superior safety profile and high efficiency, it requires multiple transfections and sophisticated mRNA engineering to minimize cellular immune responses [17]. The completely defined, vector-free nature of mRNA-derived iPSCs makes them particularly valuable for clinical applications, including the generation of cardiomyocytes for regenerative therapies [17].
Other Non-Integrating Methods
Additional non-integrating approaches include episomal plasmids, which replicate extrachromosomally and are gradually diluted through cell divisions, and adenoviral vectors, which remain episomal but typically show lower reprogramming efficiencies [17]. Minicircle DNA vectors, devoid of bacterial plasmid backbone sequences, offer enhanced transfection efficiency and prolonged transgene expression compared to conventional plasmids [17]. Protein-based reprogramming represents the ultimate in safety by directly delivering reprogramming factors, though it suffers from low efficiency and practical challenges in protein production and delivery [17].
Table 2: Comparison of Non-Integrating and DNA-Free Delivery Methods
| Method | Mechanism | Efficiency | Advantages | Disadvantages | Cardiovascular Research Utility |
|---|---|---|---|---|---|
| Sendai Virus (SeV) | Cytoplasmic RNA virus | Medium-High | Non-integrating; broad cell tropism; high efficiency | Can be difficult to clear completely; screening needed | Excellent for research and clinical applications |
| Synthetic mRNA | Direct delivery of modified mRNA | High | DNA-free; no integration risk; high efficiency | Multiple transfections; immune response concerns | Preferred for clinical-grade cardiomyocyte derivation |
| Episomal Plasmids | Extra-chromosomal replication | Medium | Non-integrating; simple production | Low efficiency; potential for integration | Research applications with verification needed |
| Adenovirus | Episomal DNA vector | Low | Non-integrating; broad tropism | Low efficiency; immune response | Limited utility due to low efficiency |
| Protein Delivery | Direct protein transduction | Low | Completely DNA-free; maximal safety | Very low efficiency; difficult protein production | Research tool with limited practical application |
Methodological Protocols
Sendai Virus Reprogramming Protocol
For cardiovascular disease modeling, the generation of iPSCs using Sendai virus vectors follows a standardized protocol. Begin with somatic cell isolation—dermal fibroblasts from skin punch biopsies or peripheral blood mononuclear cells (PBMCs) collected via venipuncture represent the most common sources [9]. PBMCs are increasingly favored for their minimally invasive collection [9].
Day 0: Plate 5×10^4 to 1×10^5 somatic cells in appropriate medium. Day 1: Infect cells with SeV vectors expressing OCT3/4, SOX2, KLF4, and c-MYC using MOI (Multiplicity of Infection) of 3-5 for each vector in serum-free medium. After 2 hours, add complete medium. Days 2-6: Change medium daily. Day 7: Transfer transduced cells onto irradiated mouse embryonic fibroblast (MEF) feeder layers or Matrigel-coated plates in human iPSC medium. Days 8-20: Continue daily medium changes. Days 21-30: Pick emerging iPSC colonies based on embryonic stem cell-like morphology and transfer to new culture vessels [19].
Monitor SeV clearance via PCR or immunostaining for viral genes. Most lines lose the viral genome by passages 8-12. For temperature-sensitive SeV variants, shifting culture temperature to 38°C can accelerate clearance [19].
mRNA Reprogramming Protocol
The mRNA reprogramming protocol requires stringent aseptic technique and daily transfections. Day 0: Plate 5×10^4 fibroblasts in 6-well plates. Days 1-16: Transfect daily with a cocktail of modified mRNAs encoding OCT4, SOX2, KLF4, c-MYC, LIN28, and NANOG using lipid-based transfection reagents. Include B18R protein in the medium to enhance mRNA stability and reduce immune responses. Days 17-30: Monitor for emerging iPSC colonies and pick for expansion [17].
Application in Cardiovascular Disease Modeling
The selection of reprogramming method directly influences the suitability of resulting iPSCs for cardiovascular disease modeling and therapeutic development. Current clinical trials using iPSC-derived cells prioritize non-integrating methods like Sendai virus and mRNA to minimize oncogenic risks [18]. In cardiovascular research, iPSCs are differentiated into cardiomyocytes (iPSC-CMs) that recapitulate disease phenotypes for conditions including arrhythmogenic disorders, heart failure, and myocardial injury [9] [3].
For instance, models of congenital arrhythmias linked to KCNQ1 mutations provide platforms for precision cardiology and drug testing [9]. However, a significant limitation remains the immature phenotype of iPSC-CMs, which resemble fetal rather than adult cardiomyocytes in their structure, metabolism, and electrophysiology [3]. Advanced engineering approaches, including 3D tissue constructs and engineered cardiac tissues (ECTs), enhance maturation and improve physiological relevance for disease modeling and drug screening applications [20].
Diagram 1: iPSC Generation and Cardiovascular Application Workflow. This diagram illustrates the complete workflow from somatic cell isolation to cardiovascular disease modeling, highlighting key viral and non-viral reprogramming methods.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of iPSC technology for cardiovascular research requires specific reagents and systems tailored to different reprogramming methods.
Table 3: Essential Research Reagents for iPSC Generation
| Reagent/System | Function | Application Notes |
|---|---|---|
| Sendai Virus Vectors (Cytotune iPS 2.0) | Delivery of OSKM factors | Temperature-sensitive variants available; monitor clearance via RT-PCR |
| mRNA Reprogramming Kit (StemRNA NP) | Modified mRNA for reprogramming | Includes immune suppressor; requires daily transfections |
| Episomal Plasmids (pCEP4-EO2S-EN2L) | Non-integrating DNA vectors | EBNA1-based system for episomal maintenance |
| Lentiviral Vectors (pSIN) | Integrating delivery | Useful for difficult-to-reprogram cells; excisable versions available |
| Yamanaka Factor Cocktail | Core reprogramming factors | OCT4, SOX2, KLF4, c-MYC (OSKM) essential for all methods |
| Matrigel/Laminin-521 | Extracellular matrix coating | Supports iPSC colony attachment and growth in feeder-free systems |
| mTeSR1/E8 Medium | Defined culture medium | Chemically formulated for feeder-free iPSC maintenance |
| Small Molecule Enhancers (e.g., Sodium Butyrate) | Epigenetic modifiers | Enhance reprogramming efficiency across multiple methods |
Diagram 2: Molecular Dynamics of Cellular Reprogramming. This diagram illustrates the key molecular and cellular events during reprogramming, highlighting the transition from early stochastic phases to late deterministic events that establish a stable pluripotent state.
The evolution of iPSC delivery methods from integrating lentiviruses to non-integrating Sendai virus and mRNA platforms reflects the field's progression toward safer, clinically applicable technologies. For cardiovascular disease modeling, the selection of reprogramming method represents a critical decision point balancing efficiency, safety, and practical considerations. While integrating methods retain utility for certain research applications, non-integrating approaches—particularly Sendai virus and synthetic mRNA—offer distinct advantages for generating clinical-grade iPSCs and their cardiovascular derivatives. Continued refinement of these technologies, coupled with advanced cardiac differentiation and tissue engineering strategies, will enhance the fidelity of iPSC-based cardiovascular disease models and accelerate the development of novel therapeutics for cardiac disorders.
The pursuit of robust in vitro models for cardiovascular disease research and drug discovery has positioned induced pluripotent stem cell (iPSC)-derived cardiomyocytes at the forefront of cardiac regenerative medicine. The differentiation of these cells mirrors embryonic heart development, a process orchestrated by precise temporal activation and inhibition of conserved signaling pathways [21] [22]. Among these, the Wnt/β-catenin, Bone Morphogenetic Protein (BMP), and Transforming Growth Factor-beta (TGF-β) pathways form a critical signaling nexus that directs iPSCs through mesoderm specification, cardiac progenitor formation, and ultimately functional cardiomyocyte maturation [23] [24]. Understanding and manipulating these pathways is not merely an academic exercise but a fundamental requirement for generating clinically relevant cardiomyocytes for disease modeling, drug screening, and therapeutic applications. This technical guide explores the mechanistic roles of these pathways and provides a practical framework for their exploitation in directing cardiac differentiation, with particular emphasis on their application within iPSC-based cardiovascular disease modeling research.
Core Signaling Pathways in Cardiac Differentiation
Wnt/β-Catenin Signaling: A Biphasic Regulator
The Wnt/β-catenin, or canonical Wnt pathway, exhibits a temporally sensitive, biphasic role in cardiac differentiation, which explains earlier seemingly contradictory findings about its function [24] [25].
- Early Activation Promotes Mesoderm Commitment: During the initial stages of differentiation, activation of the Wnt/β-catenin pathway is essential for mesoderm induction. Secreted Wnt ligands, such as Wnt3a, bind to Frizzled receptors and LRP5/6 co-receptors, preventing the destruction complex from targeting β-catenin for degradation. The stabilized β-catenin translocates to the nucleus, forming a complex with TCF/LEF transcription factors to activate target genes like Brachyury, which drives mesoderm formation [24]. Inhibition of this endogenous Wnt activity at this stage with antagonists like Dikkopf-1 (Dkk1) markedly reduces cardiac differentiation efficiency [24] [25].
- Late Inhibition Enhances Cardiogenesis: After mesoderm specification, continued Wnt/β-catenin signaling inhibits cardiac differentiation. Antagonism of the pathway during this later stage promotes the transition from cardiac progenitors to definitive cardiomyocytes [24]. This biphasic mechanism is harnessed in directed differentiation protocols, where GSK3β inhibitors like CHIR99021 are used for initial activation, followed by later inhibition using small molecules like IWP2 or IWR1 [26].
Table 1: Biphasic Role of Wnt/β-Catenin Signaling in Cardiac Differentiation
| Differentiation Phase | Signaling Status | Key Effect | Experimental Manipulation |
|---|---|---|---|
| Early (Mesoderm Induction) | Activation | Promotes formation of mesoderm and cardiac progenitors [24] [25] | Add GSK3β inhibitor (e.g., CHIR99021) or Wnt3a [26] |
| Late (Cardiac Specification) | Inhibition | Enhances differentiation of progenitors into contracting cardiomyocytes [24] | Add Wnt inhibitors (e.g., IWP2, IWR1, Dkk1) [24] |
The following diagram illustrates the Wnt/β-catenin pathway mechanism and its biphasic role in cardiac differentiation:
BMP Signaling: Driving Cardiac Progenitor Specification
BMP signaling works in concert with Wnt signaling to promote cardiac mesoderm formation and specification [23] [21].
- Mechanism: BMP ligands (e.g., BMP2, BMP4) bind to type I and type II serine/threonine kinase receptors, leading to the phosphorylation of receptor-regulated SMADs (R-SMADs: SMAD1/5/8). These phosphorylated R-SMADs form a complex with the common mediator SMAD4, which translocates to the nucleus to regulate the transcription of key cardiac genes [23].
- Key Roles in Cardiac Differentiation:
- Synergy with Activin/Nodal: BMP signaling works synergistically with Activin A (a TGF-β family member) to pattern the mesoderm toward a cardiac fate. This combination is a cornerstone of many efficient cardiomyocyte differentiation protocols [24] [25].
- Regulation of Cardiac Transcription Factors: BMP signaling directly regulates the expression of core cardiac transcription factors, including NKX2-5, ISL1, and HAND1. The SMAD complex can bind to regulatory elements of these genes, and SMAD4 interacts with NKX2-5 to promote its nuclear localization [23].
- Crosstalk with Other Pathways: A crucial crosstalk exists between BMP and Wnt/β-catenin signaling. Wnt/β-catenin signaling is required for SMAD1 activation by BMP4, indicating that Wnt signaling primes the cells for BMP activity during early mesoderm induction [24] [25].
TGF-β/Activin A Signaling: Patterning the Primitive Streak
The TGF-β pathway, particularly through Activin A and Nodal signaling, is instrumental in the initial stages of differentiation for priming pluripotent cells toward a mesendodermal lineage [24] [22].
- Mechanism: Similar to BMP signaling, Activin A binds to type I (ALK4/5/7) and type II receptors, leading to the phosphorylation of R-SMADs (SMAD2/3). The SMAD2/3-SMAD4 complex then translocates to the nucleus to direct gene expression.
- Role in Primitive Streak Patterning: Activin A signaling promotes the formation of the primitive streak and directs cells toward a definitive mesoderm fate, which is a prerequisite for cardiac specification [24]. In standard cardiac differentiation protocols, Activin A is often applied first to induce a primitive streak-like state, followed by BMP4 to drive cardiac mesoderm specification [24] [25].
The following workflow summarizes a standard protocol harnessing these pathways for cardiac differentiation:
Experimental Protocols for Directed Cardiac Differentiation
This section details a standard, highly efficient protocol for differentiating human iPSCs into cardiomyocytes by sequentially manipulating the Wnt, BMP, and TGF-β pathways, based on the activin A/BMP4 method [24] [25].
Key Reagents and Materials
Table 2: Essential Research Reagents for Cardiac Differentiation
| Reagent Category | Specific Examples | Function in Differentiation | Mechanistic Target |
|---|---|---|---|
| Cytokines/Growth Factors | Activin A [24] [25] | Initiates primitive streak patterning; induces definitive mesoderm [24] | TGF-β/Activin Pathway (SMAD2/3) |
| BMP4 [24] [25] | Specifies cardiac mesoderm; promotes cardiac progenitor formation [23] [24] | BMP Pathway (SMAD1/5/8) | |
| Wnt3a [24] [25] | Enhances mesoderm induction when added early [24] | Canonical Wnt/β-catenin Pathway | |
| Small Molecule Inhibitors/Activators | CHIR99021 [26] | GSK3β inhibitor; activates Wnt signaling for mesoderm induction [26] | Wnt/β-catenin Pathway (GSK3β) |
| IWP2 / IWR1 [24] | Wnt production/inhibition; suppresses Wnt signaling to enhance cardiogenesis [24] | Wnt/β-catenin Pathway | |
| Dkk1 [24] [25] | Secreted Wnt antagonist; inhibits early Wnt signaling and cardiogenesis [24] | Wnt/β-catenin Pathway (LRP5/6) | |
| Cell Culture Reagents | RPMI 1640 Medium [24] | Base medium for differentiation | N/A |
| B-27 Supplement (without insulin) [24] | Chemically defined supplement supporting cardiomyocyte survival | N/A | |
| Matrigel or Vitronectin [26] | Extracellular matrix for pluripotent stem cell attachment and survival | Integrin-mediated signaling |
Step-by-Step Differentiation Protocol
- Initial Cell Preparation: Culture human iPSCs to high confluence (≈80-90%) in a defined, feeder-free system, such as on Matrigel-coated plates.
- Day 0: Mesoderm Induction (Activin A): Replace the maintenance medium with differentiation medium (e.g., RPMI 1640 supplemented with B-27 minus insulin) containing Activin A (100 ng/mL). This 24-hour exposure initiates TGF-β/Activin signaling, priming cells for mesoderm formation [24] [25].
- Day 1: Cardiac Mesoderm Specification (BMP4 + Wnt Activation): Change the medium to fresh differentiation medium containing BMP4 (10 ng/mL). The protocol can be enhanced by also adding Wnt3a (10 ng/mL) or the GSK3β inhibitor CHIR99021 (2-6 µM) at this stage to further boost Wnt signaling and cardiac mesoderm specification [24] [25].
- Days 3-5: Spontaneous or Directed Wnt Inhibition: Allow the cells to develop in a basal differentiation medium without cytokines. Endogenous Wnt production typically declines in successful differentiations. Alternatively, to robustly enhance cardiomyocyte yield, add a Wnt inhibitor such as IWP2 (2 µM) or IWR1 (5 µM) between days 3 and 5 to block late Wnt signaling and promote cardiac progenitor differentiation [24].
- Days 5-14: Maturation and Beating: Continue culture in basal differentiation medium, changing the medium every 2-3 days. Spontaneously contracting clusters are typically observed between days 8 and 10.
- Day 14+: Metabolic Selection and Maintenance: From approximately day 14, replace the medium with RPMI 1640 containing B-27 Supplement (with insulin) to support long-term culture. To enrich for cardiomyocytes, a metabolic selection method using glucose-depleted lactate medium can be employed between days 10-14, which selectively supports the survival of cardiomyocytes [3].
Applications in iPSC-Based Cardiovascular Disease Modeling
The ability to generate patient-specific iPSC-derived cardiomyocytes (hiPSC-CMs) has revolutionized cardiovascular disease modeling and drug discovery, directly building upon the principles of directed differentiation [18] [3].
- Disease Modeling and Mechanism Elucidation: hiPSC-CMs allow for the reproduction of disease-specific phenotypes in a culture dish. This is particularly valuable for studying inherited cardiomyopathies (e.g., hypertrophic cardiomyopathy) and channelopathies, where patient-specific cells can reveal underlying pathological mechanisms [26]. Genome-editing tools like CRISPR-Cas9 enable the creation of isogenic control lines, confirming the causality of specific genetic mutations [26].
- Drug Discovery and Cardiotoxicity Screening: hiPSC-CMs provide a human-relevant platform for evaluating drug efficacy and safety. They can be used to assess the therapeutic potential of new compounds and are increasingly employed in preclinical cardiotoxicity screening to detect dangerous side effects like drug-induced arrhythmias, a major cause of drug attrition [3] [27]. This addresses the limitations of animal models, which often fail to predict human-specific cardiac responses due to species differences in ion channels and cardiac physiology [3].
- Clinical Trials and Regenerative Medicine: iPSC-derived cells are emerging as a promising cell-based therapy. A 2025 systematic review identified 10 published clinical studies and 22 ongoing registered trials using iPSCs to treat a range of conditions, including cardiac diseases [18]. These early studies, while small and uncontrolled, pave the way for using iPSC-derived cardiomyocytes or their secreted factors for cardiac repair in heart failure patients [18].
Current Challenges and Future Perspectives
Despite significant progress, the field faces a major hurdle: the relative immaturity of hiPSC-CMs compared to adult human cardiomyocytes [3] [27]. hiPSC-CMs typically exhibit a fetal-like phenotype, with rounded morphology, disorganized sarcomeres, absent T-tubules, immature electrophysiology, and preferential reliance on glycolytic metabolism rather than mitochondrial oxidative phosphorylation [3] [27]. This immaturity can limit their accuracy in modeling adult-onset cardiovascular diseases.
Future research is focused on developing maturation strategies that better recapitulate the adult cardiac environment. These include:
- Advanced Engineered Microenvironments: Using biomaterials like tunable hydrogels to provide physiologically relevant mechanical cues and 3D structural support that promote maturation through mechanotransduction pathways [26].
- Metabolic Manipulation: Shifting the metabolic profile of hiPSC-CMs from glycolysis to fatty acid oxidation, the primary energy source in the adult heart [3].
- Long-Term Culture and Electromechanical Stimulation: Subjecting hiPSC-CMs to prolonged culture periods and applying rhythmic electrical pacing or mechanical load to drive structural and functional maturation [26].
Overcoming the challenge of immaturity will be paramount for fully realizing the potential of hiPSC-CMs in accurately modeling cardiovascular diseases, predicting drug responses, and ultimately achieving success in regenerative therapy.
The discovery of induced pluripotent stem cells (iPSCs) marked a paradigm shift in regenerative medicine and biomedical research, offering unprecedented opportunities for disease modeling, drug discovery, and cell replacement therapies. Within cardiovascular research, iPSC technology has emerged as a particularly powerful platform for studying disease mechanisms and developing therapeutic interventions. This technical guide traces the key milestones in iPSC development from fundamental discovery to clinical-grade cell line generation, with specific emphasis on applications in cardiovascular disease modeling. The journey from basic reprogramming to clinical implementation represents a remarkable scientific achievement that has transformed our approach to understanding and treating cardiac disorders [1] [28].
The reprogramming of somatic cells to a pluripotent state fundamentally altered the stem cell research landscape, providing a patient-specific cell source without the ethical concerns associated with embryonic stem cells. For cardiovascular researchers, this technology enables generation of patient-specific cardiomyocytes that recapitulate disease phenotypes in vitro, facilitating mechanistic studies and drug screening platforms. The evolution of iPSC technology has progressed through distinct phases—from initial discovery to refinement of reprogramming methods, standardization of differentiation protocols, and ultimately development of clinical-grade cell lines compliant with regulatory standards [28].
Historical Trajectory of iPSC Technology
The foundation of iPSC technology builds upon decades of developmental biology research, with seminal discoveries paving the way for cellular reprogramming. Key historical milestones established the conceptual and technical framework that eventually enabled the generation of iPSCs:
Figure 1: Historical progression of key reprogramming milestones from early nuclear transfer to clinical application of iPSC technology.
The transformative breakthrough came in 2006 when Shinya Yamanaka's laboratory demonstrated that introducing four transcription factors—OCT4, SOX2, KLF4, and c-MYC (collectively known as OSKM)—could reprogram mouse fibroblasts into pluripotent stem cells [1] [28]. This discovery was rapidly extended to human cells in 2007 by both Yamanaka and James Thomson, generating human iPSCs from adult fibroblasts [1]. The original reprogramming approach utilized retroviral vectors that integrated into the host genome, raising concerns about potential tumorigenicity, particularly from the oncogene c-MYC [28] [29].
The mechanistic basis of reprogramming involves extensive transcriptional and epigenetic remodeling through distinct phases. Initially, somatic identity is suppressed during an initial phase, followed by stabilization of the pluripotency network. This process involves chromatin reorganization with activating histone marks (H3K4me3) enriched at pluripotency loci and reduction of repressive marks (H3K27me3). SOX2 facilitates chromatin opening and demethylation, while TET enzymes promote DNA demethylation at key regulatory genes. Signaling pathways including BMP, Wnt, and TGF-β modulate critical transitions such as the mesenchymal-to-epithelial transition (MET), which is essential for successful reprogramming [28].
Table 1: Evolution of Reprogramming Methods for iPSC Generation
| Method | Key Features | Integration Profile | Efficiency | Safety Considerations | Clinical Applicability |
|---|---|---|---|---|---|
| Retroviral Vectors | Original Yamanaka factors (OSKM) | Integrating | Low | High tumor risk, especially with c-MYC | Limited |
| Lentiviral Vectors | Can reprogram non-dividing cells | Integrating | Moderate | Insertional mutagenesis concerns | Limited |
| Sendai Virus | RNA virus, remains cytoplasmic | Non-integrating | High | Replicates in cytoplasm, eventually diluted | Good |
| Episomal Plasmids | DNA-based, Epstein-Barr origin | Non-integrating | Low to moderate | No viral elements, but low efficiency | Good |
| Synthetic mRNA | Modified to evade immune recognition | Non-integrating | Moderate | Requires repeated transfection | Excellent |
| CRISPR Activation | Targeted activation of endogenous genes | Can be integrating or non-integrating | Varies | Depends on delivery method | Emerging |
The progression toward clinical application has driven development of non-integrating reprogramming methods including Sendai virus, episomal plasmids, synthetic mRNAs, and CRISPR-based activation systems. These approaches significantly reduce the risk of insertional mutagenesis and improve the safety profile of clinical-grade iPSCs [28]. Small molecules such as CHIR99021 (a GSK3β inhibitor) and valproic acid (a histone deacetylase inhibitor) have been shown to enhance reprogramming efficiency by modulating metabolic activity and chromatin structure, further advancing the field toward robust clinical-grade cell production [28].
Progression to Clinical Applications
The translation of iPSC technology from research tool to clinical intervention has progressed rapidly, with landmark trials establishing the therapeutic potential of iPSC-derived cells. The timeline from initial discovery to clinical application has been remarkably accelerated compared to other biotechnological breakthroughs:
Table 2: Key Milestones in Clinical Translation of iPSC Technology
| Year | Milestone | Significance | Field/Application |
|---|---|---|---|
| 2006 | Discovery of mouse iPSCs | Proof-of-concept for somatic cell reprogramming | Basic Research |
| 2007 | Generation of human iPSCs | Extension to human cells, patient-specific models | Disease Modeling |
| 2013 | First human transplantation of iPSC-derived cells | iPSC-derived retinal sheets for macular degeneration | Ophthalmology |
| 2016 | First formal clinical trial of allogeneic iPSC-derived product (Cynata CYP-001) | Approval for GvHD treatment | Immunology |
| 2018 | First Parkinson's disease trial with iPSC-derived dopaminergic progenitors | Neurodegenerative disease application | Neurology |
| 2023 | Initiation of first Phase 3 iPSC trial (Cynata CYP-004) | Landmark large-scale trial for osteoarthritis | Regenerative Medicine |
| 2024 | IND approval for Eyecyte-RPE in India | Regulatory milestone for geographic atrophy | Ophthalmology |
| 2025 | Phase I/II trial of allogeneic iPSC-derived dopaminergic progenitors for Parkinson's | Demonstrated survival, dopamine production, no tumors | Neurology |
The first clinical application of iPSC-derived cells occurred in 2013 at the RIKEN Center in Kobe, Japan, led by Dr. Masayo Takahashi. This pioneering study investigated the safety of iPSC-derived retinal cell sheets in patients with age-related macular degeneration [1]. Shortly thereafter, in 2016, Cynata Therapeutics received approval to launch the first formal clinical trial of an allogeneic iPSC-derived cell product (CYP-001) for treating graft-versus-host disease (GvHD). This historic trial met its clinical endpoints and produced positive safety and efficacy data [1].
The field has now advanced to Phase 3 clinical trials, with Cynata's iPSC-derived mesenchymal stem cell product (CYP-004) being evaluated in 440 patients with osteoarthritis. This represents both the first Phase 3 clinical trial involving an iPSC-derived cell therapeutic product and the largest such trial completed to date [1]. Concurrently, numerous clinical trials are underway that do not involve transplantation but instead focus on creating and evaluating iPSC lines from specific patient populations to develop disease models [1].
Recent clinical advances include a Phase I/II trial published in 2025 reporting that allogeneic iPSC-derived dopaminergic progenitors survived transplantation, produced dopamine, and did not form tumors in Parkinson's patients [28]. Additionally, an ongoing autologous iPSC-derived dopamine neuron trial at Mass General Brigham is pioneering the use of a patient's own blood-derived iPSCs in Parkinson's disease, eliminating the need for immune suppression [28].
iPSC-Cardiomyocyte Differentiation Methodologies
The application of iPSC technology to cardiovascular disease modeling has driven development of robust cardiac differentiation protocols that generate functionally relevant cardiomyocytes. The evolution of these methods has progressed from spontaneous differentiation in embryoid bodies to highly efficient, directed differentiation protocols:
Figure 2: Evolution of cardiac differentiation protocols from initial embryoid body methods to contemporary bioreactor systems.
Initial cardiac differentiation protocols relied on spontaneous differentiation of iPSCs aggregated into embryoid bodies in KO-DMEM with 20% fetal bovine serum, yielding approximately 8% contracting embryoid bodies [30]. These were followed by co-culture systems that provided platforms for early cardiomyocyte generation without precise control of differentiation parameters, resulting in low yields and purity [30]. The field advanced significantly with the introduction of directed differentiation protocols informed by developmental biology cues, utilizing specific growth factors including activin A, FGF2, and BMP4 to improve efficiency in both embryoid body and monolayer formats [30].
The contemporary paradigm for cardiac differentiation employs sequential modulation of the Wnt/β-catenin signaling pathway, typically using small molecule inhibitors and activators. This approach begins with Wnt activation using compounds such as CHIR99021 (a GSK3β inhibitor), followed by Wnt inhibition using molecules such as IWR-1 or IWP-2 [30] [14]. This method has become the dominant approach for hiPSC-CM differentiation due to its relative simplicity and high efficiency, making it suitable for both disease modeling and therapeutic applications [14].
Recent advances have optimized suspension culture cardiac differentiation protocols to address challenges with quality, inter-batch consistency, cryopreservation, and scale that have historically reduced experimental reproducibility and clinical translation [14]. Modern bioreactor-based approaches can produce approximately 1.2 million cardiomyocytes per milliliter with ~94% purity across multiple iPSC lines. These bioreactor-differentiated cardiomyocytes (bCMs) demonstrate high viability after cryopreservation (>90%), predominantly ventricular identity, and more mature functional properties compared to standard monolayer-differentiated cardiomyocytes (mCMs) [14].
Table 3: Comparative Analysis of Cardiac Differentiation Platforms
| Parameter | Traditional Monolayer | Advanced Suspension Bioreactor |
|---|---|---|
| Scalability | Limited, linear scaling with plate area | High, efficient scaling to large volumes |
| Yield | Variable, typically lower | ~1.2E6 cells/mL, consistent |
| Purity | Variable (often <90%) | High (~94% TNNT2+) |
| Batch-to-Batch Variation | Significant | Minimal |
| Functional Maturity | Less mature phenotypes | Enhanced maturity metrics |
| Cryopreservation Recovery | Reduced viability and function | High viability (>90%) with retained function |
| Resource Requirements | Labor-intensive | More efficient at scale |
| Cost at Scale | Higher | Lower per cell |
| Clinical Translation Potential | Limited | Substantially better |
The development of stirred suspension systems represents a significant advancement in producing high-quality cardiomyocytes with reduced variability. These systems provide continuous monitoring and adjustment of temperature, O2, CO2, and pH, while constant mixing ensures optimal distribution of nutrients and differentiation factors [14]. This approach has demonstrated applicability across diverse patient-specific, gene-edited, and commercially available iPSC lines, facilitating robust cardiovascular disease modeling [14].
Technical Protocols for Cardiac Disease Modeling
Bioreactor-Based Cardiac Differentiation
An optimized suspension culture cardiac differentiation protocol developed by Fiedler et al. (2024) enables large-scale production of high-quality iPSC-derived cardiomyocytes (iPSC-CMs) with enhanced maturity and reduced batch-to-batch variability [14]. The protocol incorporates several critical advancements:
Quality-Controlled Input Cells: Establishment of master cell banks with comprehensive quality control including karyotyping and mycoplasma testing. Pluripotency marker SSEA4 is monitored by FACS, with >70% SSEA4 positivity correlating with successful differentiations (>90% TNNT2+ cells) [14].
Stirred Bioreactor System: Utilization of bioreactors that continuously monitor and adjust temperature, O2, CO2, and pH parameters throughout differentiation [14].
Small Molecule-Based Differentiation: Employment of small molecules rather than growth factors to guide differentiation, reducing costs and lot-to-lot variability [14].
Optimized Timing: Precise optimization of the initiation time point for Wnt activation based on embryoid body diameter (targeting 100μm), duration of Wnt activation (24 hours with CHIR99021), and timing of Wnt inhibition (48 hours with IWR-1 following a 24-hour gap) [14].
The protocol generates predominantly ventricular cardiomyocytes, as evidenced by high expression of ventricular markers MYH7, MYL2, and MYL3, with 83.4% of cells positive for ventricular myosin light chain (MLC2v) by flow cytometry. These cardiomyocytes demonstrate functional properties with contraction onset at differentiation day 5, earlier than monolayer-differentiated counterparts [14].
Cardiac Organoid Generation in Suspension Culture
The same research group has adapted their bioreactor protocol to generate 3D cardiac organoids entirely in suspension culture, representing the first cardiac organoid model completely generated under these conditions [14]. These organoids primarily consist of cardiomyocytes and model ventricular wall and chamber formation, similar to previously published cardiac organoid models generated in static culture. The suspension approach enables scalable production of organoids for disease modeling and drug screening applications [14].
The Scientist's Toolkit: Essential Reagents and Methodologies
Successful implementation of iPSC-based cardiovascular disease modeling requires specific reagents, tools, and methodologies carefully selected for their reliability and effectiveness:
Table 4: Essential Research Reagents for iPSC-Based Cardiac Disease Modeling
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Reprogramming Systems | Sendai Virus, Episomal Plasmids, mRNA | Somatic cell reprogramming | Non-integrating methods preferred for clinical applications |
| Cell Culture Media | Essential-8, mTeSR1, StemFlex | iPSC maintenance | Chemically defined, xeno-free formulations |
| Extracellular Matrices | Matrigel, Geltrex, Vitronectin | iPSC attachment and growth | Specific coatings support pluripotency |
| Cardiac Differentiation Agents | CHIR99021, IWR-1, BMP4, Activin A | Directed cardiac differentiation | Wnt pathway modulation is central |
| Cell Characterization Tools | Flow cytometry antibodies (TNNT2, MLC2v) | Cardiomyocyte identification and purity assessment | Quality control checkpoints |
| Functional Assay Reagents | Calcium indicators, MEA plates, Contractility sensors | Functional assessment of cardiomyocytes | Key for disease phenotyping |
| Gene Editing Tools | CRISPR/Cas9 systems, Homology-directed repair templates | Introduction or correction of disease mutations | Isogenic control generation |
| Cryopreservation Solutions | DMSO-based cryomedium with controlled rate freezing | Cell storage and banking | Critical for maintaining viability |
The integration of artificial intelligence and machine learning methodologies represents an emerging tool in the iPSC workflow. These technologies enable automated colony morphology classification, differentiation outcome prediction, and enhanced standardization in iPSC manufacturing [28]. AI-guided approaches are increasingly applied to optimize culture conditions for large-scale iPSC production and analyze genetic and omics data to uncover biological patterns relevant to personalized medicine applications [29].
CRISPR-Cas9 genome editing has become an indispensable tool for creating precise disease models and conducting mechanistic studies. In cardiovascular research, CRISPR enables introduction of specific disease-associated mutations into healthy iPSC lines or correction of mutations in patient-derived iPSCs, generating isogenic control lines that are genetically identical except for the mutation of interest [28] [31]. This approach controls for genetic background variability and strengthens conclusions about genotype-phenotype relationships.
The progression from the initial discovery of iPSCs to the development of clinical-grade cell lines represents a remarkable scientific achievement that has fundamentally transformed cardiovascular research and regenerative medicine. Key milestones including the refinement of reprogramming methods, standardization of cardiac differentiation protocols, implementation of quality control measures, and execution of pioneering clinical trials have established iPSC technology as a cornerstone of modern biomedical science. For cardiovascular disease modeling specifically, advances in bioreactor-based differentiation and organoid generation have enabled production of high-quality, clinically relevant cardiomyocytes at scales necessary for drug screening and therapeutic applications. Despite substantial progress, challenges remain in achieving full maturation of iPSC-cardiomyocytes, ensuring genomic stability, and scaling manufacturing processes to meet clinical demand. The ongoing integration of enabling technologies such as CRISPR gene editing, single-cell omics, and artificial intelligence promises to further advance the field toward robust clinical implementation and personalized cardiovascular medicine.
Advanced hiPSC-CM Applications: Disease Modeling and Drug Screening
The advent of human induced pluripotent stem cells (iPSCs) has revolutionized the study of inherited cardiac channelopathies by providing a uniquely human, patient-specific model system that bridges the gap between traditional animal models and human pathophysiology [32] [33]. These cells, generated through the reprogramming of somatic cells such as dermal fibroblasts or peripheral blood cells, possess the critical advantage of maintaining the complete genetic background of the donor, including all disease-causing mutations and genetic modifiers [34] [33]. For complex arrhythmia syndromes such as Long QT Syndrome (LQTS) and Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), iPSC-derived cardiomyocytes (iPSC-CMs) enable researchers to move beyond mere phenotypic recapitulation to mechanistic interrogation of disease processes in a controlled human cellular environment [32]. This technological advancement is particularly valuable for cardiovascular disease modeling because it bypasses the limitations of primary human cardiomyocytes, which are difficult to obtain and expand, while also overcoming the species-specific differences that often render animal models inadequate for predicting human cardiac electrophysiology and drug responses [33] [35].
Within the broader context of cardiovascular disease modeling research, iPSC systems now occupy a critical role in bridging basic discovery with translational applications [32]. They have enabled unprecedented insight into human-specific disease mechanisms, including the role of splice variants, transcriptional regulation, and mitochondrial stress in arrhythmogenesis [32]. Furthermore, the ability to integrate iPSC technology with genome editing tools such as CRISPR/Cas9 has empowered researchers to establish definitive causal links between genetic variants and cellular phenotypes, particularly for variants of uncertain significance (VUS) that present diagnostic challenges in clinical practice [33]. As the field progresses, advances in directed differentiation now permit chamber-specific cardiac cell generation, allowing for more precise atrial and ventricular disease modeling and revealing critical cell-cell interactions that contribute to arrhythmogenesis [32]. These developments position iPSC technology as an indispensable component of the modern cardiovascular researcher's toolkit, contributing significantly to personalizing care and advancing therapeutics in inherited arrhythmic syndromes [32].
Disease Modeling: Long QT Syndrome and ARVC
Long QT Syndrome (LQTS) Modeling with iPSC-CMs
Long QT Syndrome represents a group of inherited cardiac arrhythmogenic disorders characterized by prolonged ventricular repolarization, reflected in a lengthened QT interval on the surface electrocardiogram, and an increased risk of potentially fatal ventricular tachyarrhythmias [33] [35]. iPSC-based modeling has been particularly instrumental in advancing our understanding of specific LQTS subtypes, with research revealing novel disease mechanisms beyond simple ion channel loss-of-function.
For LQT2, caused by mutations in KCNH2 encoding the hERG potassium channel, iPSC studies have uncovered the significant role of alternative splicing in disease pathogenesis [33]. Research using patient-specific iPSCs harboring the H70R mutation within the hERG N-terminal PAS domain demonstrated not only impaired channel trafficking of the hERG1a isoform but also a compensatory increase in hERG1b mRNA levels and an altered hERG1b/hERG1a ratio [33]. This altered ratio leads to faster deactivation of IKr and longer repolarization times, revealing a dual pathogenic mechanism that expands our understanding beyond simple haploinsufficiency [33]. Similarly, studies of Timothy Syndrome (LQT8), caused by mutations in CACNA1C encoding the L-type calcium channel, have utilized patient-specific iPSCs to identify novel therapeutic approaches. Yazawa et al. demonstrated that roscovitine, which enhances voltage-dependent inactivation of CaV1.2, could restore electrical and Ca2+ signaling properties of patient-specific iPSC-CMs, with subsequent mechanistic studies revealing that this effect occurs through partial inhibition of CDK5 to regulate CaV1.2 [33].
iPSC models have also proven invaluable for interrogating variants of uncertain significance (VUS) in LQTS genes. Terrenoire et al. investigated familial LQTS using patient-specific iPSCs harboring two VUS from both SCN5A (F1473C) and KCNH2 (K897T) [33]. Patch-clamp recordings revealed that enhanced late Na+ current (INaL) driven by the SCN5A polymorphism was primarily responsible for the LQT3 phenotype, while the KCNH2-K897T variant showed normal current density, enabling more accurate variant classification according to ACMG guidelines [33].
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) Modeling with iPSC-CMs
Arrhythmogenic Right Ventricular Cardiomyopathy is characterized by progressive replacement of ventricular myocardium with fibrofatty tissue, ventricular arrhythmias, and an increased risk of sudden cardiac death [33]. While traditionally considered a structural disorder, iPSC models have revealed important electrophysiological alterations that contribute to its arrhythmogenic potential.
Mutations in the LMNA gene, encoding nuclear envelope proteins lamin A and lamin C, are implicated in a form of arrhythmogenic cardiomyopathy that shares features with ARVC [33]. Patient-specific iPSC-derived cardiomyocytes modeling LMNA-related cardiomyopathy (heterozygous A388fs) have demonstrated how mutant lamin A induces accelerated degradation of SIRT1, leading to increased oxidative stress, elevated reactive oxygen species (ROS), and mitochondrial dysfunction [33]. This ROS-activated Ca2+/calmodulin-dependent protein kinase II (CaMKII) mediates sarcoplasmic reticulum Ca2+ leak, promoting arrhythmogenesis and uncovering a novel mechanism underlying laminopathies [33]. Importantly, elevated ROS creates a feedforward loop that increases accumulation of SUN1, which further disrupts nuclear envelope structure [33].
iPSC models have also revealed how transcription factor mutations can contribute to arrhythmogenesis in ARVC. Bersell et al. reported that a TBX5 G145R variant associated with arrhythmogenic pathology reduced transcriptional activity, downregulated SCN5A and platelet-derived growth factor receptor α (PDGFR), blunted peak INa and enhanced INaL [33]. Similarly, the LMNA K219T mutation functions as a genetic modifier by enhancing binding of mutant lamin A/C proteins to the SCN5A promoter in patient-derived iPSC-CMs, resulting in histone repression at SCN5A and concomitant reduction in NaV1.5 expression [33]. These findings emphasize that arrhythmogenesis in cardiomyopathies can occur through transcriptional regulation mechanisms that are best studied in human cellular systems.
Table 1: Key Electrophysiological Characteristics of iPSC-CMs in Channelopathy Modeling
| Parameter | Long QT Syndrome Models | ARVC/LMNA Models | Control iPSC-CMs |
|---|---|---|---|
| Action Potential Duration | Significantly prolonged [33] [35] | Prolonged [33] | Variable but shorter [35] |
| Major Ionic Current Alterations | Reduced IKr (LQT2) [33]; Enhanced late INa (LQT3) [33] | Reduced peak INa; Enhanced late INa [33] | Immature profile with low IK1 [35] |
| Calcium Handling | Abnormal Ca2+ transients (Timothy Syndrome) [33] | SR Ca2+ leak via CaMKII activation [33] | Immature SR function [35] |
| Arrhythmogenic Triggers | Early afterdepolarizations [35] | Delayed afterdepolarizations [33] | Spontaneous automaticity [35] |
| Response to Pharmacological Intervention | Roscovitine restores Ca2+ signaling (Timothy Syndrome) [33] | SIRT1 manipulation reduces arrhythmogenesis [33] | Variable based on maturity |
Experimental Design and Methodologies
iPSC Generation and Quality Control
The generation of human iPSCs begins with the selection of an appropriate somatic cell source. Dermal fibroblasts obtained via skin punch biopsy and peripheral blood cells collected through routine venipuncture represent the most common sources, with each offering distinct advantages [34]. Blood cells provide easier donor recruitment and the potential to utilize previously stored samples, while fibroblasts may carry a lower mutational burden in older donors [34]. The reprogramming process typically employs non-integrating methods such as Sendai virus, episomal plasmid vectors, or mRNA transfection to deliver the Yamanaka factors (Oct3/4, Sox2, Klf4, and c-Myc), minimizing the risk of genomic integration while efficiently inducing pluripotency [34].
Following successful reprogramming, rigorous quality control measures are essential to ensure iPSC line suitability for cardiac channelopathy modeling. These assessments include karyotyping to verify genomic integrity, pluripotency validation through marker expression (e.g., Nanog, SSEA-4, Tra-1-60/Tra-1-81), and teratoma formation assays to demonstrate differentiation potential across all three germ layers [34]. Additionally, researchers should confirm the absence of residual reprogramming vectors through PCR analysis and verify the retention of patient-specific mutations via Sanger sequencing or other genomic techniques [34].
Cardiac Differentiation Protocols
Efficient differentiation of iPSCs into cardiomyocytes typically employs modulated Wnt signaling pathway activation followed by inhibition, recapitulating developmental cardiacogenesis [35]. Most protocols begin with the formation of embryoid bodies or monolayer cultures that are treated with activators of the Wnt pathway (e.g., CHIR99021, a GSK-3β inhibitor) during the initial stages of differentiation, followed by Wnt inhibition (e.g., IWP2 or IWR1) to promote cardiac mesoderm specification and subsequent cardiomyocyte differentiation [35]. The resulting heterogeneous population of cardiomyocytes typically includes ventricular-like, atrial-like, and nodal-like cells, which can be further enriched using metabolic selection techniques based on the preferential lactate metabolism of cardiomyocytes versus glucose-dependent pluripotent stem cells [35].
For chamber-specific modeling, particularly relevant for ARVC which predominantly affects the right ventricle, researchers have developed directed differentiation protocols that generate atrial or ventricular subtypes through modulation of retinoic acid signaling during cardiac specification [32]. The efficiency of cardiac differentiation and the resulting cellular heterogeneity should be quantitatively assessed through flow cytometry for cardiac-specific markers (e.g., cTnT, α-actinin), electrophysiological characterization of action potential morphology, and transcriptomic analysis of chamber-specific markers (e.g., MYL2 for ventricular identity) [32] [35].
Functional Characterization of iPSC-CMs
Comprehensive electrophysiological assessment represents a cornerstone of channelopathy modeling with iPSC-CMs. Patch clamp electrophysiology remains the gold standard for evaluating action potential parameters (duration, morphology, upstroke velocity) and specific ionic currents (INa, ICaL, IKr, IKs, IK1) that are typically disrupted in channelopathies [33] [35]. For high-throughput screening, multi-electrode array (MEA) systems enable non-invasive recording of field potentials from spontaneously beating monolayers, providing information on repolarization duration (analogous to QT interval) and arrhythmia incidence [33] [35].
Calcium handling abnormalities, which contribute significantly to arrhythmogenesis in both LQTS and ARVC, can be assessed using ratiometric calcium dyes (e.g., Fura-2) or genetically-encoded calcium indicators in combination with live-cell fluorescence imaging [33] [35]. These techniques reveal important parameters including calcium transient amplitude, duration, decay kinetics, and the presence of spontaneous calcium release events that trigger delayed afterdepolarizations [33]. For structural assessment, particularly in ARVC models, immunocytochemistry and confocal microscopy evaluate sarcomeric organization (α-actinin), gap junction distribution (Cx43), and nuclear integrity (lamin A/C) [33].
Table 2: Essential Methodologies for iPSC-CM Functional Characterization
| Methodology | Key Parameters Assessed | Application in Channelopathy Modeling | Technical Considerations |
|---|---|---|---|
| Patch Clamp Electrophysiology | Action potential duration, Ionic current density, Channel kinetics | Gold standard for quantifying repolarization abnormalities in LQTS; Identifying specific ion channel defects | Low-throughput; Technically demanding; Requires specialized equipment |
| Multi-Electrode Array (MEA) | Field potential duration, Beat rate, Arrhythmia incidence | High-throughput assessment of proarrhythmic phenotypes; Drug screening | Non-invasive; Limited spatial resolution; Indirect measurement of repolarization |
| Calcium Imaging | Calcium transient duration, Spontaneous calcium release events, SR calcium load | Assessing abnormal calcium handling in CPVT and ARVC models | Can combine with pharmacological challenges; Requires fluorescent indicators |
| Immunocytochemistry | Sarcomeric organization, Connexin distribution, Nuclear morphology | Evaluating structural changes in ARVC; Assessing cardiomyocyte maturity | Qualitative and quantitative analysis possible; Dependent on antibody specificity |
Signaling Pathways and Molecular Mechanisms
The molecular pathophysiology of cardiac channelopathies involves complex interactions between ion channels, regulatory proteins, and signaling networks. iPSC-based studies have been instrumental in elucidating these mechanisms, particularly through the integration of patient-specific cells with genome editing technologies.
In LQT2, the pathogenic mechanism extends beyond simple loss-of-function of the hERG channel. Research using iPSC-CMs has revealed that mutations in the N-terminal PAS domain of KCNH2 disrupt normal channel trafficking to the membrane while also altering the relative expression ratio of hERG1a and hERG1b splice variants [33]. This altered ratio favors the hERG1b isoform, which demonstrates faster deactivation kinetics, further compromising repolarization reserve and prolonging action potential duration [33]. The following diagram illustrates this pathogenic mechanism:
In ARVC related to LMNA mutations, iPSC models have uncovered a novel pathway connecting nuclear envelope defects to electrophysiological instability. Mutant lamin A induces accelerated degradation of SIRT1, leading to mitochondrial dysfunction and increased reactive oxygen species (ROS) production [33]. ROS subsequently activates Ca2+/calmodulin-dependent protein kinase II (CaMKII), which mediates sarcoplasmic reticulum calcium leak through ryanodine receptor hyperphosphorylation, promoting delayed afterdepolarizations and triggered arrhythmias [33]. Additionally, elevated ROS creates a feedforward loop by increasing accumulation of SUN1, further disrupting nuclear envelope integrity [33]. This pathway is illustrated below:
Therapeutic Applications and Drug Development
iPSC-CMs have emerged as a powerful platform for drug screening and therapeutic development for cardiac channelopathies, enabling both mechanism-specific drug discovery and personalized assessment of drug efficacy and safety [32] [36]. The capacity to generate unlimited numbers of patient-specific human cells that reproduce hallmark disease features in culture makes iPSCs uniquely qualified for pharmacological applications, including the discovery of mechanism-specific therapeutics, evaluation of safety and efficacy in a human context before clinical trials, and stratification of patients for clinical trials based on cellular drug responses [36].
In LQTS modeling, iPSC-CMs have enabled testing of mutation-specific therapies. For Timothy Syndrome (LQT8), roscovitine and its analogs have been shown to enhance voltage-dependent inactivation of mutant CaV1.2 channels, restoring normal electrical and calcium signaling properties in patient-specific iPSC-CMs [33]. Mechanistic studies revealed that this effect occurs through partial inhibition of CDK5, regulating CaV1.2 and identifying CDK5 as a potential therapeutic target [33]. Similarly, in JLN syndrome patient-derived iPSC lines with complex homozygous loss of KCNQ1, disruption of the hERG1 PAS domain increased IKr, shortened action potential durations, and decreased arrhythmogenic events, suggesting a novel therapeutic approach for this severe arrhythmia syndrome [33].
For ARVC related to LMNA mutations, iPSC models have identified potential interventions targeting the newly discovered pathogenic pathway. Manipulation of SIRT1 in iPSC-CMs interrupted the cycle of ROS production, CaMKII activation, and RYR2-induced arrhythmogenesis, as well as nuclear envelope deformation [33]. Similarly, antioxidant approaches targeting mitochondrial ROS production show promise in mitigating the arrhythmogenic phenotype in these models [33].
Beyond specific therapeutic compounds, iPSC-CMs have become invaluable for preclinical cardiac safety assessment, particularly for evaluating drug-induced arrhythmogenesis. The comprehensive electrophysiological profiling possible with iPSC-CMs allows researchers to identify compound effects on multiple ion currents and action potential parameters simultaneously, providing a more integrated assessment of proarrhythmic potential than traditional single-channel assays [36]. This application is particularly important for identifying off-target effects that might exacerbate underlying channelopathies in susceptible patients.
Technical Challenges and Limitations
Despite their significant advantages, iPSC-based models of cardiac channelopathies face several important technical challenges that must be considered when interpreting experimental results. The most significant limitation remains the immature phenotype of iPSC-derived cardiomyocytes, which more closely resemble fetal rather than adult human cardiomyocytes in their structural and functional properties [35]. This immaturity manifests in several critical aspects: spontaneous automaticity due to incomplete suppression of HCN channel activity and elevated funny current (I_f) density; depolarized resting membrane potentials resulting from reduced expression of the inward rectifier potassium channel (IK1); and abnormal calcium handling due to underdeveloped sarcoplasmic reticulum and t-tubule networks [35]. These electrophysiological differences can complicate the modeling of adult-onset channelopathies and potentially mask or alter disease phenotypes.
Cellular heterogeneity represents another significant challenge in iPSC-based modeling. Standard differentiation protocols typically generate mixed populations of ventricular-like, atrial-like, and nodal-like cardiomyocytes, introducing electrical dispersion that can confound functional analyses [35]. This heterogeneity, while reflective of early cardiac development, creates a substrate for reentrant arrhythmias and complicates the interpretation of disease-specific phenotypes [35]. Single-cell RNA sequencing and metabolic selection strategies are being employed to address this limitation, but achieving pure populations of specific cardiomyocyte subtypes remains technically challenging [35].
From a methodological standpoint, the field currently lacks standardized protocols for iPSC generation, cardiac differentiation, and functional assessment, making cross-study comparisons difficult [34]. Additionally, issues of scalability and reproducibility can present obstacles for high-throughput drug screening applications [36]. Ongoing efforts to address these limitations include the development of advanced maturation strategies using prolonged culture, mechanical loading, electrical stimulation, and 3D tissue engineering; integration of multi-cellular systems to better recapitulate the cardiac microenvironment; and the establishment of standardized quality control metrics for assessing iPSC-CM maturity and functional integrity [32] [35].
Table 3: Key Research Reagent Solutions for iPSC-Based Channelopathy Modeling
| Reagent/Resource | Function | Application Examples | Technical Notes |
|---|---|---|---|
| Reprogramming Methods | Generation of iPSCs from somatic cells | Sendai virus (non-integrating), episomal vectors, mRNA transfection [34] | Non-integrating methods preferred for reduced genomic alteration risk [34] |
| Cardiac Differentiation Kits | Directed differentiation of iPSCs to cardiomyocytes | Commercial kits available based on Wnt modulation; Metabolic selection reagents [35] | Protocol optimization often required for different iPSC lines; Quality varies between lots |
| Genome Editing Tools | Introduction or correction of disease-associated variants | CRISPR/Cas9 for isogenic control generation; VUS characterization [33] | Essential for establishing causality; Requires careful off-target effect assessment |
| Electrophysiology Platforms | Functional characterization of iPSC-CMs | Patch clamp systems; Multi-electrode arrays; Voltage-sensitive dyes [33] [35] | MEA allows higher throughput; Patch clamp provides gold standard data quality |
| Maturation Enhancers | Promotion of adult-like cardiomyocyte phenotype | Engineered substrates; Hormone treatment; Electrical stimulation systems [32] [35] | Critical for modeling adult-onset diseases; Multiple approaches often combined |
| Cell Lines and Biobanks | Source of disease-specific and control iPSCs | Arrhythmia-specific lines available through repositories like Coriell; Consortium biobanks [34] | Essential controls include isogenic lines and lines from healthy donors |
The field of iPSC-based modeling of cardiac channelopathies continues to evolve rapidly, with several promising directions emerging that will further enhance the utility and translational relevance of these models. Advances in tissue engineering are enabling the creation of more physiologically relevant 3D cardiac microtissues that better recapitulate the structural and functional complexity of the native myocardium [32]. These engineered tissues demonstrate improved maturation, enhanced sarcomeric organization, and more normal electrophysiological properties compared to 2D cultures, potentially addressing the current limitations associated with iPSC-CM immaturity [32] [35]. Additionally, the integration of iPSC technology with computational modeling approaches is enabling the development of increasingly sophisticated in silico representations of cardiac electrophysiology that can predict emergent tissue-level behaviors from cellular data [32].
From a therapeutic perspective, iPSC-based models are poised to play an expanding role in personalized medicine approaches for cardiac channelopathies. The ability to generate patient-specific cardiomyocytes enables preclinical testing of multiple therapeutic options to identify the most effective strategy for individual patients, particularly for those with rare or novel mutations [36]. Furthermore, the combination of iPSC technology with high-content screening and machine learning approaches promises to accelerate the discovery of novel therapeutic compounds that target specific molecular defects in LQTS, ARVC, and other inherited arrhythmia syndromes [36].
In conclusion, iPSC-based modeling has fundamentally transformed our approach to studying inherited cardiac channelopathies like Long QT Syndrome and ARVC, providing unprecedented insight into human-specific disease mechanisms and creating new opportunities for therapeutic development. While technical challenges remain, ongoing advances in differentiation protocols, maturation strategies, and tissue engineering are rapidly addressing these limitations. As part of the broader thesis on iPSC applications in cardiovascular disease modeling, this platform represents a critical bridge between basic discovery and clinical translation, holding significant promise for personalizing care and improving outcomes for patients with these potentially life-threatening genetic disorders.
Induced pluripotent stem cells (iPSCs) have emerged as a transformative technology for modeling human cardiovascular diseases in vitro. By enabling the generation of patient-specific cardiomyocytes, iPSCs provide a unique platform to study the pathological mechanisms of hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) while preserving the patient's complete genetic background [37] [38]. This approach addresses critical limitations of traditional models, including species-specific differences in cardiac physiology observed in animal models and the limited availability of human primary cardiomyocytes for research [39] [3]. The "disease-in-a-dish" paradigm powered by iPSC technology allows researchers to recapitulate disease phenotypes de novo, facilitating the investigation of genotype-phenotype relationships and the development of targeted therapeutic interventions [38] [12].
The application of iPSC-derived cardiomyocytes (iPSC-CMs) is particularly valuable for studying inherited cardiomyopathies like HCM and DCM, which are caused by mutations in genes encoding cardiac sarcomeric proteins, cytoskeletal elements, and ion channels [40] [38]. iPSC-CMs capture the individual's genetic makeup, including modifiers that may influence disease penetrance and expressivity, thereby offering unprecedented opportunities for personalized disease modeling and drug screening [37] [41]. As the field progresses, advanced three-dimensional (3D) models including engineered heart tissues, microtissues, and organoids are further enhancing the physiological relevance of these in vitro systems by better mimicking the native cardiac microenvironment [41].
Disease Pathogenesis and Genetic Foundations
Hypertrophic Cardiomyopathy (HCM)
HCM is the most common inherited cardiac disorder, with an estimated prevalence of 1 in 500 individuals [40] [38]. It is characterized by left ventricular hypertrophy unexplained by secondary causes, myofibrillar disarray, diastolic dysfunction, and an increased risk of arrhythmias [40] [42]. Approximately half of all HCM cases are familial with autosomal dominant inheritance, primarily caused by mutations in genes encoding sarcomeric proteins [40]. The genetic landscape of HCM is dominated by mutations in MYBPC3 (encoding myosin-binding protein C) and MYH7 (encoding β-myosin heavy chain), which together account for approximately 50% of familial cases [40] [38]. Other less frequently mutated genes include TNNT2, TNNI3, TPM1, ACTC1, MYL2, MYL3, and CSRP3, collectively accounting for a smaller proportion of cases [40].
The pathogenesis of HCM involves multiple interconnected mechanisms triggered by sarcomeric gene mutations. These include impaired contractile function, altered calcium handling, disrupted energy metabolism, and activation of hypertrophic signaling pathways [40] [3]. The initial compensatory hypertrophy eventually leads to maladaptive remodeling, characterized by fibrosis, microvascular dysfunction, and electrophysiological abnormalities that create a substrate for arrhythmias [42].
Dilated Cardiomyopathy (DCM)
DCM is characterized by systolic dysfunction and ventricular dilation, often progressing to heart failure [38] [41]. With an estimated prevalence of 1 in 2500, DCM represents a leading indication for cardiac transplantation [38]. While acquired forms exist due to viral myocarditis, toxic exposures, or autoimmune processes, genetic factors play a significant role in 30-50% of idiopathic DCM cases [38] [41]. The genetic architecture of DCM is more heterogeneous than HCM, involving mutations in genes encoding a diverse array of proteins including cytoskeletal elements, nuclear envelope proteins, and sarcomeric components [41].
The most commonly implicated gene in DCM is TTN, encoding the giant sarcomeric protein titin, which accounts for approximately 25% of familial cases [38] [41]. Other important disease genes include LMNA (encoding lamin A/C), MYH7, MYH6, TNNT2, SCN5A, and DES (encoding desmin) [38] [41]. LMNA mutations are particularly noteworthy for their association with high risk of malignant arrhythmias and rapid disease progression [38]. The pathogenic mechanisms in DCM involve impaired force transmission, disrupted nuclear integrity, altered ion channel function, and aberrant signal transduction, ultimately leading to cardiomyocyte dysfunction, apoptosis, and ventricular remodeling [41].
Table 1: Major Genetic Mutations in HCM and DCM
| Cardiomyopathy | Gene | Protein | Frequency | Primary Functional Consequences |
|---|---|---|---|---|
| HCM | MYBPC3 | Myosin-binding protein C | ~20% | Impaired sarcomere regulation |
| HCM | MYH7 | β-myosin heavy chain | ~15% | Altered contractile force generation |
| HCM | TNNT2 | Cardiac troponin T | ~2% | Disrupted calcium sensitivity |
| HCM | TNNI3 | Cardiac troponin I | ~2% | Abnormal thin filament regulation |
| DCM | TTN | Titin | ~25% | Impaired sarcomere assembly & passive tension |
| DCM | LMNA | Lamin A/C | ~5-8% | Nuclear envelope instability, arrhythmogenesis |
| DCM | MYH7 | β-myosin heavy chain | ~4% | Reduced contractile force |
| DCM | SCNA5 | Cardiac sodium channel | ~2% | Arrhythmia susceptibility |
iPSC Generation and Cardiomyocyte Differentiation
Somatic Cell Reprogramming to iPSCs
The generation of iPSCs begins with the reprogramming of somatic cells into a pluripotent state through the forced expression of specific transcription factors. The original method described by Takahashi and Yamanaka utilized four factors: OCT4, SOX2, KLF4, and c-MYC (collectively known as OSKM or Yamanaka factors) [12]. An alternative combination developed by Thomson and colleagues uses OCT4, SOX2, NANOG, and LIN28 [12]. These reprogramming factors work collaboratively to reactivate the pluripotency network and erase the somatic epigenetic memory, ultimately establishing a self-sustaining pluripotent state [12].
Multiple reprogramming methods have been developed, each with distinct advantages and limitations. Early approaches used integrating retroviral or lentiviral vectors, which raise safety concerns due to potential insertional mutagenesis and transgene reactivation [40] [12]. More recent non-integrating methods include Sendai virus, episomal plasmids, synthetic mRNA, and small molecule-based approaches, which eliminate the risk of genomic integration [40] [12]. The choice of reprogramming method depends on the specific application, with non-integrating methods preferred for clinical applications and disease modeling due to their superior safety profile [12].
Various somatic cell sources can be used for reprogramming, including dermal fibroblasts, peripheral blood mononuclear cells, and urinary epithelial cells [40] [38]. The selection of starting material involves considerations of accessibility, reprogramming efficiency, and culture requirements. Peripheral blood mononuclear cells have gained popularity due to the minimal invasiveness of collection and relatively high reprogramming efficiency [38].
Cardiomyocyte Differentiation Protocols
Efficient differentiation of iPSCs into cardiomyocytes is achieved through stage-specific manipulation of developmental signaling pathways, primarily Wnt/β-catenin signaling [38] [3]. The most widely used protocols mimic cardiac development in vivo by sequentially directing cells through mesoderm formation, cardiac mesoderm specification, and cardiomyocyte maturation [3].
The basal differentiation medium typically consists of RPMI 1640 supplemented with B-27 or defined lipid concentrates. Key small molecules used to direct cardiac differentiation include:
- CHIR99021: A GSK-3β inhibitor that activates Wnt signaling to induce mesoderm formation (typically used at 4-8 μM for 24-48 hours) [3]
- IWP-2/IWP-4: Wnt inhibitors that block porcupine-mediated Wnt secretion, applied after mesoderm formation to promote cardiac specification (typically used at 2-5 μM) [3]
- Ascorbic acid: Enhances cardiomyocyte differentiation and viability [3]
The differentiation process typically yields contracting cardiomyocytes within 7-12 days, with efficiency rates often exceeding 80-90% in optimized protocols [3]. Metabolic selection using glucose-depleted, lactate-containing media can further enrich the cardiomyocyte population to >95% purity by exploiting the preferential utilization of lactate over glucose in cardiomyocytes [3].
Figure 1: Workflow for iPSC Generation and Cardiac Differentiation
Advanced 3D Cardiac Models
Two-dimensional iPSC-CM cultures have provided fundamental insights into disease mechanisms but face limitations due to their structural simplicity and immature phenotype. To better recapitulate the native myocardial environment, researchers have developed sophisticated 3D models that promote cellular maturation and enable the study of tissue-level pathophysiology [41]. These advanced platforms incorporate multiple cell types, biomechanical cues, and complex cell-cell interactions that more accurately mimic the in vivo cardiac tissue architecture and function [41].
Engineered Heart Tissues (EHTs)
EHTs are 3D constructs typically generated by embedding iPSC-CMs alone or in combination with other cardiac cells (fibroblasts, endothelial cells) within hydrogels composed of natural extracellular matrix proteins such as collagen type I and Matrigel [41]. The mixture is cast into custom-designed molds that promote uniaxial alignment and subjected to static or dynamic mechanical loading. EHTs have been successfully used to model both HCM and DCM, demonstrating disease-specific phenotypes including hypercontractility and impaired relaxation in HCM and reduced contractile force in DCM [41]. A key advantage of EHTs is their ability to measure isometric force generation and contractile kinetics, providing functional readouts that closely mirror cardiac performance in vivo [41].
Cardiac Microtissues and Spheroids
Cardiac microtissues (MTs) are self-assembled 3D aggregates that can be generated through various methods including hanging drop, ultra-low attachment plates, or forced aggregation techniques [41]. These models often incorporate multiple cell types in ratios resembling native myocardium (typically 70% iPSC-CMs, 15% cardiac fibroblasts, and 15% endothelial cells) to better recapitulate cellular crosstalk and paracrine signaling [41]. When derived from HCM patient-specific iPSCs, MTs exhibit structural disorganization and arrhythmic behavior, mirroring key features of the disease [41]. Similarly, DCM-derived MTs show impaired contractile function and altered electrophysiological properties [41].
Cardiac Organoids
Cardiac organoids represent the most advanced in vitro models, featuring self-organizing structures that mimic aspects of native heart development and organization [41]. Recent protocols have enabled the generation of "epicardioids" containing both myocardial and epicardial components, which have proven valuable for modeling HCM-associated fibrotic responses and arrhythmias [41]. These complex models more accurately recapitulate the multicellular pathogenesis of cardiomyopathies, including the interplay between cardiomyocytes and non-myocyte cells that drive structural remodeling and dysfunction [41].
Table 2: Comparison of 3D Cardiac Model Systems
| Model Type | Key Components | Advantages | Limitations | Applications in HCM/DCM |
|---|---|---|---|---|
| Engineered Heart Tissues (EHTs) | iPSC-CMs, collagen/Matrigel, mechanical loading | Functional force measurements, aligned structure | Limited cellular complexity, specialized equipment required | Hypercontractility assessment (HCM), contractile deficit (DCM) |
| Cardiac Microtissues | iPSC-CMs, cardiac fibroblasts, endothelial cells | Multicellular interactions, self-assembly, medium throughput | Limited structural organization, small size | Arrhythmia modeling, fibrotic responses |
| Cardiac Organoids | Multiple cardiac lineages, self-patterning | Developmental recapitulation, complex cellular crosstalk | Protocol variability, high technical expertise required | Fibrosis modeling, developmental pathogenesis |
Phenotypic Characterization of Disease Models
Structural and Morphological Analysis
Comprehensive characterization of iPSC-CM models involves multidisciplinary approaches to assess disease-specific phenotypic alterations. In HCM models, structural analysis typically reveals cellular hypertrophy, with increased cell size and elevated actinin-positive area compared to healthy controls [40] [42]. Sarcomeric organization is frequently disrupted, characterized by myofibrillar disarray and irregular Z-disc patterning [40] [3]. These structural abnormalities are often accompanied by molecular features of pathological hypertrophy, including re-expression of fetal genes such as atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) [40].
DCM models typically exhibit sarcomeric disorganization and reduced cell density, reflecting the dilated phenotype and systolic dysfunction characteristic of the disease [41]. At the ultrastructural level, electron microscopy often reveals mitochondrial abnormalities and disrupted intercellular junctions in both HCM and DCM models [40] [41]. Advanced imaging techniques including confocal microscopy, super-resolution imaging, and live-cell imaging enable quantitative analysis of these morphological changes and their dynamic evolution over time [3].
Functional Assessment
Functional characterization of iPSC-CM models encompasses contractility, electrophysiology, and calcium handling assessments. HCM iPSC-CMs typically demonstrate hypercontractility at the single-cell level, with increased contraction velocity and amplitude [40] [42]. In contrast, DCM models show reduced contractile function with slowed contraction kinetics and decreased force generation [41]. These measurements can be performed using video-based edge detection systems, atomic force microscopy, or traction force microscopy for 2D cultures, and force transducers for 3D EHTs [3] [41].
Electrophysiological assessment using multi-electrode arrays or patch clamp techniques reveals arrhythmogenic phenotypes in both HCM and DCM models. HCM iPSC-CMs frequently exhibit action potential prolongation, early afterdepolarizations, and increased arrhythmia susceptibility following electrical stimulation [40] [42]. Calcium handling is typically impaired in both diseases, with HCM models showing calcium handling abnormalities including slowed calcium decay kinetics and increased calcium spark frequency [40] [3]. DCM models often demonstrate reduced calcium transient amplitude and abnormal sarcoplasmic reticulum calcium loading [41].
Molecular Profiling
Molecular analyses provide insights into the pathogenic mechanisms underlying disease phenotypes. Transcriptomic profiling through RNA sequencing reveals disease-specific gene expression signatures, including metabolic alterations, stress pathway activation, and fetal gene reprogramming [40] [41]. Proteomic approaches identify changes in protein expression, post-translational modifications, and sarcomeric protein stoichiometry [3]. Metabolic characterization typically demonstrates a shift from fatty acid oxidation to glycolytic metabolism in HCM models, recapitulating the fetal metabolic profile observed in patients [40] [3].
Table 3: Key Phenotypic Features in HCM and DCM iPSC Models
| Parameter | HCM Phenotype | DCM Phenotype | Assessment Methods |
|---|---|---|---|
| Cell Morphology | Increased cell size, hypertrophy | Normal or decreased cell size, structural dilation Microscopy, flow cytometry | |
| Sarcomere Organization | Myofibrillar disarray, irregular Z-discs | Sarcomeric disorganization, reduced density | Immunostaining (α-actinin, TM), electron microscopy |
| Contractile Function | Hypercontractility, increased force | Reduced contraction, decreased force | Video motion detection, AFM, EHT force measurements |
| Electrophysiology | AP prolongation, EADs, arrhythmias | AP shortening, DADs, conduction slowing | Patch clamp, MEA, optical mapping |
| Calcium Handling | Slowed decay, increased spark frequency | Reduced amplitude, abnormal SR load | Calcium imaging (Fluo-4, Fura-2) |
| Gene Expression | Fetal gene re-expression (ANF, BNP) | Metabolic pathway alterations | RNA-seq, qPCR, nanostring |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for iPSC-Based Cardiomyopathy Modeling
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Reprogramming Factors | OCT4, SOX2, KLF4, c-MYC (OSKM) | Somatic cell reprogramming to pluripotency | Use non-integrating delivery methods (episomal, mRNA) for clinical applications |
| Cardiomyocyte Differentiation Inducers | CHIR99021, IWP-2/IWP-4, BMP4, Ascorbic acid | Directed cardiac differentiation from iPSCs | Optimize concentration and timing for specific iPSC lines |
| Metabolic Selection Agents | Lactate, glucose-free media | Cardiomyocyte purification | Apply at day 7-12 of differentiation for 3-5 days |
| Maturation Enhancers | T3 (triiodothyronine), IGF-1, Dexamethasone, Fatty acids | Promote structural and functional maturation | Combine multiple factors for synergistic effects |
| Fluorescent Calcium Indicators | Fluo-4, Fura-2, Rhod-2 | Calcium transient measurements | Use pluronic acid for dye loading; validate loading efficiency |
| Contractility Assessment Tools | Edge detection software, AFM, traction force microscopy | Quantitative analysis of contraction | Maintain consistent temperature and pH during measurements |
| 3D Matrix Components | Collagen I, Matrigel, fibrin | Scaffold for engineered tissues | Optimize stiffness to match native myocardium (~10-15 kPa) |
| Electrophysiology Tools | Multi-electrode arrays, patch clamp systems | Action potential and conduction analysis | Maintain physiological temperature during recordings |
Experimental Workflows for Disease Modeling
HCM Modeling Protocol
A comprehensive experimental workflow for HCM modeling involves multiple stages, beginning with the generation of patient-specific iPSCs from somatic cells (typically dermal fibroblasts or peripheral blood mononuclear cells) using non-integrating reprogramming methods [40] [38]. Once iPSC lines are established and validated, they are differentiated into cardiomyocytes using established protocols with stage-specific modulation of Wnt signaling [40] [3]. For isogenic control generation, CRISPR/Cas9 genome editing is employed to correct the disease-causing mutation in patient iPSCs or introduce specific mutations into healthy iPSCs [40] [41].
Phenotypic characterization should include assessment of hypertrophy markers, sarcomeric organization, contractile function, electrophysiological properties, and calcium handling [40] [42]. For 3D models, EHTs or microtissues are generated using patient-specific and isogenic control iPSC-CMs, with functional assessment including force measurements, arrhythmia susceptibility testing, and pharmacological profiling [41]. High-content imaging and multi-parameter analyses enable quantitative comparison of disease phenotypes and evaluation of therapeutic interventions [3] [41].
DCM Modeling Protocol
DCM modeling follows a similar overall workflow but with specific focus on DCM-relevant phenotypes. Following iPSC generation and cardiomyocyte differentiation, DCM models are characterized by assessment of cell size, sarcomeric structure, contractile function, and apoptotic susceptibility [41]. Electrophysiological analysis should include detailed evaluation of conduction velocity and arrhythmia triggers, particularly for models with LMNA or SCN5A mutations [38] [41].
3D models of DCM should incorporate mechanical loading conditions that mimic ventricular wall stress, as mechanical overload is a key driver of DCM progression [41]. Functional assessment includes measurement of twitch force, contraction and relaxation kinetics, and response to inotropic agents [41]. Molecular analyses should focus on pathways regulating cytoskeletal integrity, nuclear envelope function, and stress response signaling [38] [41].
Figure 2: Experimental Workflow for Cardiomyopathy Modeling
Current Challenges and Future Perspectives
Despite significant advances, several challenges remain in the field of iPSC-based cardiomyopathy modeling. A primary limitation is the immature phenotype of iPSC-CMs, which more closely resemble fetal rather than adult cardiomyocytes in terms of their structural, metabolic, electrophysiological, and contractile properties [3]. Current maturation strategies include prolonged culture duration, metabolic manipulation, electrical stimulation, mechanical loading, 3D culture, and neurohormonal stimulation, but achieving full adult maturity remains elusive [3] [41].
Technical challenges include inter-line variability, standardization of differentiation protocols, and cost-effective scaling for high-throughput applications [18] [3]. The field is addressing these limitations through the development of defined culture conditions, quality control standards, and automated platforms for consistent large-scale production of iPSC-CMs [3].
Future directions include the integration of multi-omics approaches, microphysiological systems, and artificial intelligence to enhance the predictive power of iPSC-based models [37] [38]. There is also growing emphasis on developing more complex models that incorporate immune cells, vascular networks, and innervation to better recapitulate the tissue microenvironment [41]. As these technologies mature, iPSC-based cardiomyopathy models are poised to play an increasingly important role in drug discovery, safety pharmacology, and the development of personalized therapeutic approaches for HCM and DCM patients [37] [38] [41].
The clinical translation of iPSC technology is already underway, with recent systematic reviews identifying 10 published clinical studies and 22 ongoing registered trials utilizing iPSCs for various conditions, including cardiac diseases [18]. While most current studies are small and uncontrolled, they provide valuable preliminary data on safety and feasibility that will guide future larger-scale trials [18]. Standardization of iPSC-derived product characterization and outcome reporting will be essential for accelerating the clinical adoption of iPSC-based therapies for cardiomyopathies [18].
Human induced pluripotent stem cells (iPSCs) have emerged as a transformative platform for cardiovascular disease modeling, drug discovery, and toxicity screening. By capturing patient-specific genetic information, iPSCs enable the generation of limitless supplies of cardiomyocytes (iPSC-CMs) that recapitulate key aspects of human cardiac physiology and pathology [34] [10]. This technology addresses critical limitations of traditional models, including species-specific differences in cardiac biology that undermine the predictive value of animal studies and the limited availability of primary human cardiomyocytes for large-scale screening [3]. The field has progressed significantly since the pioneering work of Takahashi and Yamanaka, who first demonstrated that somatic cells could be reprogrammed to a pluripotent state using defined transcription factors [34] [43].
Current applications of iPSC technology in cardiovascular research span disease modeling, personalized drug testing, cardiotoxicity screening, and regenerative therapy development. A recent scoping review of clinical studies and registered trials identified 10 published clinical studies and 22 ongoing registered trials utilizing iPSCs to treat various conditions, including cardiac diseases, ocular disorders, and cancer [18]. This review highlights the emerging clinical promise of iPSC-based approaches while noting that most current evidence comes from small, uncontrolled studies, with only 115 total patients treated across published studies [18]. Despite these limitations in clinical translation, iPSC-CMs have become established tools for preclinical cardiotoxicity assessment, particularly through initiatives like the Comprehensive in vitro Proarrhythmia Assay (CiPA), which leverages human iPSC-CMs to enhance cardiac safety evaluation beyond traditional hERG channel testing [44] [45].
Current Landscape and Clinical Translation
The clinical application of iPSC-based therapies is advancing cautiously but steadily. A systematic scoping review published in 2025 analyzed the landscape of iPSC clinical studies and found investigations spanning cardiac conditions, ocular disorders, cancer, graft-versus-host disease, and platelet production for transfusion [18]. The same review noted that while published studies were predominantly small and uncontrolled, the field is poised for significant impact on clinical care as standardization improves [18].
Table 1: Published Clinical Studies Utilizing iPSCs (as of January 2025)
| Medical Area | Number of Studies | Patient Population | Phase of Development |
|---|---|---|---|
| Cardiac Conditions | Multiple ongoing | Patients with heart failure | Early-phase trials |
| Ocular Disorders | Several published | Patients with retinal diseases | Phase I/II studies |
| Cancer | Multiple approaches | Cancer patients | Early-phase trials |
| Graft-versus-Host Disease | Limited studies | Patients undergoing transplantation | Early investigations |
| Platelet Transfusion | Proof-of-concept | Patients requiring transfusion | Early-phase trials |
The translation of iPSC technology from bench to bedside faces several challenges, including manufacturing complexities, regulatory hurdles, and concerns about potential tumorigenicity from residual undifferentiated cells [18]. Manufacturing iPSC-derived cells is a multi-step process involving donor cell procurement, transfection with reprogramming factors, and redifferentiation into specific cell types using specific growth factors and small molecules [18]. Additionally, the reprogramming process can introduce unintentional genetic changes, increasing tumor risk, while transplanting residual undifferentiated iPSCs could lead to uncontrolled cell proliferation [18]. Despite these challenges, ongoing clinical trials continue to explore the therapeutic potential of iPSCs across multiple disease areas.
Cardiovascular Disease Modeling with iPSCs
Genetic Cardiovascular Diseases
iPSCs offer unprecedented opportunities for modeling inherited cardiovascular conditions through their ability to capture patient-specific genetic backgrounds. Researchers have successfully used iPSC-CMs to model various cardiomyopathies, channelopathies, and structural heart diseases [34] [10]. By combining patient-derived iPSCs with genome editing tools like CRISPR-Cas9, isogenic control lines can be created, allowing direct comparison between diseased and genetically corrected cells from the same genetic background [26]. This approach enables researchers to definitively link specific genetic variants to disease phenotypes while controlling for confounding genetic factors.
Initial progress has been made in using iPSCs to better understand cardiomyopathies, rhythm disorders, valvular and vascular disorders, and metabolic risk factors for ischemic heart disease [34]. For example, iPSC-CMs carrying hypertrophic cardiomyopathy (HCM) mutations have recapitulated key disease features, including cellular hypertrophy, sarcomeric disorganization, and altered calcium handling [26] [10]. Similarly, iPSC-CMs from patients with long QT syndrome have demonstrated prolonged action potential duration and abnormal electrophysiological properties that mirror the clinical phenotype [10].
Acquired Cardiovascular Diseases
Beyond inherited conditions, iPSC-CMs can model acquired cardiovascular diseases, including drug-induced cardiotoxicity and heart failure with various etiologies. These models are particularly valuable for studying the interplay between genetic predisposition and environmental factors in cardiovascular pathology [46] [10]. For instance, iPSC-CMs have been used to model doxorubicin-induced cardiotoxicity, with cells from different donors exhibiting variable susceptibility that mirrors the clinical spectrum of this serious adverse drug reaction [46].
Cardiac mechanobiology—how cardiac cells sense and respond to mechanical stimuli—plays a crucial role in both physiological adaptation and pathological remodeling [26]. iPSC-CMs have enabled unprecedented investigation into these processes, revealing how mechanical stress influences disease progression through integrin- and cadherin-mediated signaling pathways [26]. However, a significant limitation remains the relatively immature phenotype of standard iPSC-CMs, which more closely resemble fetal than adult cardiomyocytes [26] [3]. This immaturity potentially limits their accuracy in modeling adult-onset cardiovascular diseases.
Drug-Induced Cardiotoxicity Screening
Comprehensive Cardiotoxicity Assessment
Drug-induced cardiotoxicity remains a major challenge in pharmaceutical development, leading to serious cardiovascular complications and the withdrawal of previously approved drugs [44] [45]. Traditional cardiotoxicity screening has primarily focused on QT interval prolongation and arrhythmia risk, particularly Torsades de Pointes [45]. However, this narrow focus often fails to capture the complex interplay among multiple ion channels or detect early manifestations of chronic cardiotoxicity [45].
The CiPA initiative represents a significant advancement by integrating multi-ion channel assays, human iPSC-CM models, and in silico simulations into a unified framework [44] [45]. This approach more comprehensively evaluates proarrhythmic risk beyond just hERG channel blockade. Recent studies have validated CiPA-qualified human iPSC-CMs for toxicity testing, demonstrating their ability to correctly classify drugs based on arrhythmia risk [44]. In one investigation, 28 drugs were tested on hiPSC-CMs using electric field potential measurements, with results aligning with established CiPA classifications [44]. Notably, the model identified droperidol and domperidone—classified as intermediate-risk compounds—as high-risk, consistent with previous clinical observations [44].
Table 2: Cardiotoxicity Assessment Platforms Using iPSC-CMs
| Platform | Key Measurements | Advantages | Limitations |
|---|---|---|---|
| Microelectrode Array (MEA) | Field potential duration, beating rate | Non-invasive, multi-site recording | Sparse electrode configuration in traditional systems |
| Ultra-high-density CMOS-MEA | Excitation origins, conduction velocity, propagation area | High spatial resolution (236,880 electrodes) | Technically complex, specialized equipment needed |
| Impedance-based Systems | Beat rate, amplitude, cell viability | Detects morphological changes and cytotoxicity | Does not directly measure electrophysiological parameters |
| Calcium Transient Imaging | Calcium handling, arrhythmias | Evaluates excitation-contraction coupling | Requires fluorescent dyes or indicators |
Advanced Technologies for Cardiotoxicity Detection
Recent technological innovations have significantly enhanced the resolution and predictive accuracy of cardiotoxicity screening using iPSC-CMs. Ultra-high-density complementary metal-oxide-semiconductor microelectrode arrays (UHD-CMOS-MEAs) containing 236,880 electrodes distributed across a 5.9 × 5.5 mm active area now enable detailed mapping of action potential propagation with near single-cell resolution [45]. This system can extract novel electrophysiological endpoints, including the number and spatial variability of excitation origins, conduction velocity, and propagation area [45].
This high-resolution platform has demonstrated superior sensitivity in detecting both acute and chronic cardiotoxicity. For instance, chronic cardiotoxicity induced by low-dose doxorubicin (0.03 μM) was detected within 24 hours—earlier and at lower concentrations than previously reported—based on significant reductions in conduction velocity and propagation area [45]. Multivariate analysis incorporating both conventional and novel endpoints enabled accurate classification of drug mechanisms under acute conditions, highlighting the platform's potential for mechanism-aware cardiotoxicity profiling [45].
Donor-Specific Variability in Cardiotoxicity
A significant advantage of iPSC-CMs is their ability to capture interindividual differences in drug sensitivity, enabling more personalized cardiotoxicity prediction [46]. This donor-specific variability mirrors the clinical reality where patients exhibit different susceptibilities to drug-induced cardiovascular complications. Proof-of-concept studies have demonstrated that iPSC-CMs generated from multiple donors show greater line-to-line variability than intraindividual variability in impedance cytotoxicity and transcriptome assays [46].
In one investigation, doxorubicin-induced cardiotoxicity was studied using 20 iPSC-CM lines generated from donors with varying cardiovascular phenotypes [46]. The variable and dose-dependent cytotoxic responses of these iPSC-CMs resembled the spectrum of responses observed in clinical practice and largely replicated reported mechanisms of toxicity [46]. By categorizing iPSC-CMs into resistant and sensitive cell lines based on their phenotypic responses to doxorubicin, researchers found that the sensitivity of donor-specific iPSC-CMs may predict in vivo cardiotoxicity risk [46]. Furthermore, transcriptomic analysis identified differentially expressed genes between doxorubicin-resistant and doxorubicin-sensitive iPSC-CMs, suggesting potential biomarkers for predicting individual susceptibility [46].
Experimental Protocols and Methodologies
iPSC-CM Culture and Maintenance
Standardized protocols for iPSC-CM culture and maintenance are essential for reproducible cardiotoxicity testing. Commercially available iPSC-CMs, such as iCell Cardiomyocytes (FUJIFILM Cellular Dynamics, Inc.), are typically handled according to supplier instructions [45]. Generally, cryopreserved cells are thawed in a 37°C water bath and resuspended in specialized thawing medium to prepare a cell suspension at a final concentration of 1 × 10^7 cells/ml [45]. For microelectrode array assays, cells are seeded directly onto fibronectin-coated electrode areas at densities of approximately 100,000 cells per chip [45]. After allowing cell attachment for 1 hour at 37°C, maintenance medium is gently added. The culture medium is typically replaced 24 hours after seeding, with half the volume exchanged regularly thereafter [45]. For cardiotoxicity testing, iPSC-CMs are usually used between days 7 and 10 in vitro (DIV), when spontaneous beating activity is consistently observed [45].
Cardiotoxicity Assessment Using Microelectrode Arrays
The following protocol details cardiotoxicity assessment using microelectrode array systems:
- Chip Preparation: MEA chips are cleaned with Tergazyme solution, rinsed with distilled water, treated with 70% ethanol, air-dried, and UV-sterilized [45].
- Surface Coating: Chips are coated overnight at 4°C with diluted Type-C collagen, air-dried, then treated with fibronectin solution (50 μg/ml in PBS) and incubated at 37°C for 1 hour [45].
- Cell Plating: iPSC-CMs are seeded onto the fibronectin-coated electrode area and incubated at 37°C for 1 hour to allow attachment before adding maintenance medium [45].
- Pharmacological Testing: Test compounds are dissolved in culture medium containing 0.1% DMSO and administered cumulatively at increasing concentrations [45]. Measurements begin 30 minutes after compound administration [45].
- Data Acquisition: Field potential parameters are recorded, including beat rate, field potential duration (FPD), amplitude, and conduction velocity [45]. For UHD-CMOS-MEA systems, additional parameters include number and spatial variability of excitation origins and propagation area [45].
- Data Analysis: Compound effects are analyzed based on concentration-dependent changes in measured parameters, with comparison to established reference compounds [45].
Donor-Specific Cardiotoxicity Screening Protocol
For assessing interindividual variability in cardiotoxicity susceptibility:
- iPSC Generation: Generate iPSC lines from multiple donors using non-integrating methods such as Sendai virus or episomal vectors [34] [46].
- Cardiomyocyte Differentiation: Differentiate iPSCs into cardiomyocytes using standardized protocols, such as Wnt pathway modulation [46] [3].
- Quality Control: Verify pluripotency markers for iPSCs and cardiac markers (e.g., cTnT, MLC-2v) for iPSC-CMs [34] [46].
- Phenotypic Screening: Expose iPSC-CMs from different donors to test compounds across a range of concentrations [46].
- Endpoint Assessment: Measure multiple endpoints, including:
- Data Stratification: Classify donors as sensitive or resistant based on predefined thresholds of response [46].
- Biomarker Identification: Compare transcriptomic and proteomic profiles between sensitive and resistant lines to identify potential predictive biomarkers [46].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for iPSC-Based Cardiotoxicity Screening
| Reagent/Platform | Function | Examples/Suppliers |
|---|---|---|
| iPSC-CMs | Patient-specific cardiotoxicity testing | iCell Cardiomyocytes (Fujifilm CDI), Cor.4U (Ncardia) |
| Microelectrode Arrays | Extracellular electrophysiology recording | Multiwell MEA systems (Axion Biosystems, Maxwell Biosystems) |
| UHD-CMOS-MEA | High-resolution field potential imaging | 236,880-electrode platform (MaxOne) |
| Impedance Systems | Cell viability and beating analysis | xCELLigence RTCA Cardio (Agilent) |
| Calcium Imaging Dyes | Calcium transient measurement | Fluo-4, Cal-520, Rhod-2 |
| Differentiation Kits | Cardiomyocyte generation from iPSCs | Commercial differentiation kits (StemCell Technologies) |
| Extracellular Matrix | Cell attachment and maturation | Fibronectin, Matrigel, laminin-521 |
| Ion Channel Modulators | Reference compounds for validation | E-4031 (hERG blocker), nifedipine (Ca2+ channel blocker) |
Artificial Intelligence and Advanced Analytics
The integration of artificial intelligence, particularly machine learning and deep learning, has significantly advanced iPSC technology by refining cell classification, monitoring functionality, and analyzing genetic data [43]. A 2024 systematic scoping review identified 79 studies applying AI to iPSC research, with applications spanning cell type classification, assessment of disease-specific phenotypes in iPSC-derived cells, and drug screening [43]. The United States emerged as the leading contributor in this field, accounting for 39.2% of identified studies [43].
AI approaches have demonstrated particular utility in addressing the challenge of differentiation efficiency in iPSC protocols. One recent study developed an early prediction system for muscle stem cell differentiation efficiency using phase contrast imaging and machine learning [47]. By applying Fast Fourier Transform for feature extraction and random forest classification, the system could predict final differentiation efficiency approximately 50 days before the end of the induction period—less than half the total protocol duration [47]. This approach enabled a 43.7% reduction in the defective sample rate and a 72% increase in the number of high-quality samples [47].
For cardiotoxicity assessment, AI algorithms can analyze complex, high-dimensional data from UHD-CMOS-MEA systems to identify subtle patterns indicative of toxicological risk [43] [45]. Multivariate analysis incorporating both conventional and novel electrophysiological endpoints has enabled accurate classification of drug mechanisms, potentially reducing false positives and false negatives in cardiotoxicity screening [45]. These advances create a foundation for future improvements in iPSC-based technologies for drug discovery and safety assessment.
Future Perspectives and Challenges
Despite significant progress, several challenges remain in optimizing iPSC technology for drug discovery and toxicity screening. The immature phenotype of conventional iPSC-CMs continues to limit their physiological relevance, particularly for modeling adult-onset cardiovascular diseases [26] [3]. Current maturation strategies include:
- Engineered substrates with physiological stiffness to promote structural maturation
- Metabolic manipulation to shift from glycolytic to oxidative metabolism
- Electrical and mechanical stimulation to mimic in vivo conditions
- 3D tissue engineering to enhance cell-cell and cell-matrix interactions
- Non-myocyte co-culture to replicate cardiac microenvironment [26] [3]
Standardization remains another critical challenge. Considerable variability exists in study design, medical conditions examined, cell sources used for iPSC generation, and specific iPSC-derived products across different research groups [18]. This heterogeneity complicates comparisons between studies and may delay definitive understanding of iPSC-based therapy safety and efficacy [18]. Moving forward, standardized study protocols and adherence to consistent cell product characterization criteria will be essential for accelerating clinical translation [18].
The field is also evolving toward more complex model systems that better recapitulate cardiac physiology. Engineered heart tissues and heart-on-a-chip platforms that integrate multiple cell types and incorporate mechanical and electrical stimulation offer more physiologically relevant contexts for drug testing [26]. These advanced systems may bridge the gap between traditional 2D cultures and in vivo conditions, potentially improving the predictive accuracy of preclinical cardiotoxicity screening.
As these technologies mature, iPSC-based approaches are poised to transform early drug discovery and safety assessment, enabling more human-relevant screening, personalized toxicity prediction, and ultimately, more effective and safer therapeutics for patients with cardiovascular disease.
The advent of induced pluripotent stem cell (iPSC) technology has revolutionized cardiovascular disease modeling, enabling the generation of patient-specific cardiomyocytes for research and therapeutic applications. While traditional two-dimensional (2D) cultures have provided valuable insights, they fundamentally lack the complex cellular ecosystems and three-dimensional architecture of native human heart tissue. This limitation has driven the development of sophisticated three-dimensional (3D) models—specifically engineered heart tissues and cardiac organoids—that more faithfully recapitulate the structural and functional properties of the human myocardium. These advanced models represent a critical bridge between conventional cell culture and human physiology, offering unprecedented opportunities for studying disease mechanisms, screening drug candidates, and developing regenerative therapies. By incorporating essential features such as multi-cellularity, physiologically relevant extracellular matrix, and emergent tissue-level functionality, 3D cardiac models are transforming our approach to understanding and treating cardiovascular diseases [48] [49].
The Evolution from 2D to 3D Cardiac Modeling
Limitations of Traditional 2D Cultures
Traditional 2D cultures of iPSC-derived cardiomyocytes (iPSC-CMs), while valuable, exhibit significant limitations that restrict their translational relevance. These models typically demonstrate immature electrophysiological properties, disorganized sarcomere architecture, and altered metabolic activity more reminiscent of fetal than adult cardiomyocytes. Crucially, they lack the cell-cell and cell-matrix interactions that govern cardiac development, maturation, and disease progression in vivo. The absence of physiological tissue stiffness and three-dimensional cytoarchitecture in 2D systems results in aberrant signaling and mechanotransduction, further limiting their predictive value for human biology and drug responses [48] [49].
Advantages of 3D Cardiac Models
Three-dimensional cardiac models address these limitations by providing a more physiologically relevant microenvironment that promotes enhanced structural maturation, improved electromechanical coupling, and adult-like metabolic activity. The spatial constraints and mechanical forces inherent to 3D cultures drive the organization of aligned sarcomeres, development of mature excitation-contraction coupling, and establishment of tissue-scale synchronous beating. Furthermore, 3D models enable the incorporation of multiple cell types—including cardiac fibroblasts, endothelial cells, and immune cells—in appropriate spatial orientations, recapitulating the cellular crosstalk essential for normal cardiac function and disease pathogenesis [50] [48] [49].
Table 1: Comparative Analysis of 2D vs 3D Cardiac Culture Systems
| Parameter | 2D Culture | 3D Engineered Tissue | 3D Cardiac Organoid |
|---|---|---|---|
| Structural Maturity | Disorganized sarcomeres; fetal phenotype | Aligned sarcomeres; improved Z-disc development | Self-organizing structure; embryonic heart features |
| Functional Properties | Irregular, weak contractions; immature action potentials | Synchronous, strong contractions; mature electrophysiology | Spontaneous beating; chamber-like compartmentalization |
| Cellular Complexity | Typically mono-culture | Can incorporate endothelial cells and fibroblasts | Multiple cardiac lineages; emergent tissue patterning |
| Drug Response | Often fails to predict clinical toxicity | Improved predictive value for contractility effects | Potential for developmental toxicity screening |
| Throughput | High | Medium | Low to medium |
| Applications | Basic research, initial toxicity screening | Disease modeling, drug discovery, safety pharmacology | Developmental biology, disease modeling, regeneration studies |
Methodological Approaches for Generating 3D Cardiac Tissues
Scaffold-Based Engineered Heart Tissues
Scaffold-based approaches utilize biomaterials to provide structural support and biochemical cues that guide tissue assembly and maturation. These systems employ natural polymers such as fibrin, collagen, and Matrigel, or synthetic hydrogels like PEG-fibrinogen, which offer tunable mechanical properties and degradation rates. The biomaterial serves as a temporary extracellular matrix that supports cell adhesion, spreading, and tissue formation while allowing for the application of biophysical cues such as mechanical stretch and electrical stimulation that further promote maturation. A notable example combines alginate with PEG-fibrinogen to create 3D bioprinted cardiac tissues containing both iPSC-CMs and human umbilical vein endothelial cells (HUVECs) to model vascularized myocardium [50] [51].
Scaffold-Free Cardiac Organoids
Cardiac organoids represent a scaffold-free approach that leverages the inherent self-organizing capacity of stem cells to form complex 3D structures resembling embryonic heart development. These models are generated through controlled differentiation protocols in suspension culture, often using low-adhesion plates or bioreactors to allow for spontaneous aggregation and tissue patterning. The resulting organoids contain multiple cardiac cell types arranged in spatially organized patterns that recapitulate aspects of early heart development, including the formation of chamber-like structures and vascular networks. Recent advances have demonstrated the generation of human heart organoids (hHOs) using a two-step Wnt signaling modulation strategy, which produces structures containing cardiomyocytes, endothelial cells, and cardiac fibroblasts [14] [49].
Advanced Bioprinting Strategies
3D bioprinting technologies enable the precise spatial patterning of cells and biomaterials to create cardiac tissues with defined architecture and composition. Innovative approaches include:
- Microfluidic printing heads for simultaneous deposition of multiple bioinks containing different cell types
- Embedded 3D printing within support matrices to enable freeform fabrication of complex structures
- Light-based stereolithography for creating microscale features that guide cellular alignment These techniques allow for the creation of cardiac micro-tissues with anisotropic architecture that mimics the aligned organization of native myocardium, significantly enhancing structural and functional maturity compared to 2D cultures [52] [53] [51].
Signaling Pathways Governing Cardiac Differentiation and Maturation
The successful generation of functional cardiac tissues from iPSCs requires precise temporal control of key developmental signaling pathways. The Wnt/β-catenin pathway plays a particularly critical role, with sequential activation and inhibition driving cardiac specification. Initial Wnt activation promotes mesoderm commitment, while subsequent inhibition facilitates cardiac progenitor formation and cardiomyocyte differentiation. Additional pathways including BMP, TGF-β, and Notch signaling further refine cardiac patterning and maturation [54].
Diagram Title: Signaling Pathway for Cardiac Differentiation
Experimental Protocols for Generating 3D Cardiac Models
Stirred Suspension Bioreactor Protocol for iPSC-Cardiomyocytes
This optimized protocol generates high-purity ventricular cardiomyocytes with enhanced maturity and reduced batch-to-batch variability [14]:
Starting Material Preparation: Begin with quality-controlled iPSC master cell banks characterized by karyotyping and mycoplasma testing. Ensure pluripotency marker SSEA4 expression exceeds 70% by FACS analysis.
Embryoid Body Formation: Dissociate iPSCs to single cells and transfer to stirred suspension bioreactor. Culture in appropriate medium with continuous monitoring and adjustment of temperature, O₂, CO₂, and pH.
Mesoderm Induction: When embryoid bodies reach 100μm diameter (typically 24 hours), add 7μM CHIR99021 (Wnt activator) for 24 hours.
Cardiac Specification: After 24-hour gap, add 5μM IWR-1 (Wnt inhibitor) for 48 hours to direct cardiac lineage commitment.
Cardiomyocyte Maturation: Continue culture in cardiac maturation medium with metabolic selection if desired. Spontaneous contractions typically begin at differentiation day 5.
Harvest and Cryopreservation: Harvest cardiomyocytes at differentiation day 15-30 using enzymatic dissociation. Implement controlled freeze protocols to maintain viability (>90% post-thaw).
Table 2: Quantitative Outcomes of Bioreactor Cardiac Differentiation Protocol [14]
| Parameter | Value | Notes |
|---|---|---|
| Cell Yield | ~1.21 million cells/mL | Approximately 2.4 hiPSC-CMs per input iPSC |
| Purity (TNNT2+) | ~94% | Consistent across multiple iPSC lines |
| Viability Post-Thaw | >90% | With optimized cryopreservation protocol |
| Ventricular Identity | 83.4% MLC2v+ | Predominantly ventricular cardiomyocytes |
| Onset of Contraction | Differentiation day 5 | Earlier than monolayer differentiation (day 7) |
| Batch-to-Batch Variation | Low | Coefficient of variation <10% for key markers |
3D Bioprinting Protocol for Cardiac Micro-Tissues
This rapid protocol creates aligned cardiac micro-tissues suitable for high-throughput drug screening [50] [53]:
Cell Preparation: Differentiate iPSCs to cardiomyocytes using established protocols. At differentiation day 15±1, harvest iPSC-CMs using 0.25% trypsin solution.
Bioink Formulation: Resuspend cell pellet (5.0×10⁷–1.5×10⁸ cells) in 100μL culture medium with ROCK inhibitor. Mix with 0.9mL GelXA Laminink-521 bioink at 37°C.
Bioprinting Parameters: Load cell-bioink mixture into 3mL syringe fitted with 22G nozzle. Print at 25°C with extrusion pressure of 18-30 kPa. Create constructs with 3 vertical layers (4.0mm × 4.0mm × 0.6mm).
Crosslinking: Expose constructs to 365nm ultraviolet light for 15 seconds, followed by immersion in 50mM CaCl₂ crosslinking solution for 1 minute.
Culture and Maturation: Maintain constructs in RPMI/B-27 medium with 20% FBS. Change medium every 3 days. Spontaneous contractions typically begin within 3-7 days.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Cardiac Tissue Engineering
| Reagent/Material | Function | Example Applications |
|---|---|---|
| GelXA Laminink-521 | Bioink with defined laminin composition | 3D bioprinting of cardiac constructs [50] |
| CHIR99021 | GSK-3β inhibitor; activates Wnt signaling | Mesoderm induction in cardiac differentiation [14] [54] |
| IWR-1/IWP-2 | Wnt pathway inhibitors | Cardiac specification after mesoderm induction [14] [54] |
| PEG-Fibrinogen | Synthetic-natural hybrid hydrogel | Scaffold for bioprinted cardiac tissues [51] |
| Alginate | Temporary structural template | Support during bioprinting process [51] |
| ROCK inhibitor (Y-27632) | Enhances cell survival after dissociation | Used during cell passaging and bioprinting [50] |
| Matrigel | Basement membrane extract | Traditional 3D culture; being replaced by defined matrices [49] |
| Lactate | Metabolic selection agent | Enriches cardiomyocytes by eliminating non-cardiac cells [54] |
Applications in Disease Modeling and Drug Development
Modeling Cardiovascular Disorders
3D cardiac tissues have demonstrated particular utility in modeling both genetic and acquired cardiovascular diseases. For genetic conditions such as long QT syndrome, 3D models recapitulate the disease phenotype more faithfully than 2D cultures, exhibiting prolonged action potentials and arrhythmogenic susceptibility. For structural heart diseases like hypoplastic left heart syndrome (HLHS), patient-specific iPSC-derived cardiac tissues enable investigation of disease mechanisms and evaluation of potential interventions. The enhanced structural organization of 3D models is essential for studying sarcomeric cardiomyopathies, as the spatial arrangement of contractile proteins directly influences disease pathogenesis [50] [49].
Cardiotoxicity Screening and Drug Discovery
The pharmaceutical industry has increasingly adopted 3D cardiac models for preclinical safety assessment, addressing the limitation of traditional systems that fail to predict clinical cardiotoxicity. Recent approaches combine functional measurements (e.g., multi-electrode array analysis of electrophysiology) with high-content imaging of subcellular morphology to create comprehensive cardiotoxicity profiles. This integrated approach has demonstrated 76% accuracy in classifying known cardiotoxic compounds, significantly outperforming single-parameter assays. The enhanced predictive value of 3D models is particularly valuable for detecting diverse toxicity mechanisms, including structural damage, mitochondrial dysfunction, and arrhythmogenesis [55].
Regenerative Medicine and Tissue Repair
Cardiac tissues engineered from human iPSCs show tremendous promise for myocardial repair following injury. Preclinical studies have demonstrated the ability of 3D cardiac patches to integrate with host myocardium, improve contractile function, and promote angiogenesis. The incorporation of vascular networks within engineered tissues enhances their survival and functional integration following implantation. Additionally, 3D cardiac organoids serve as powerful tools for investigating cardiac development and endogenous regenerative mechanisms, potentially informing novel therapeutic strategies for heart regeneration [49] [51].
Current Challenges and Future Directions
Despite significant advances, several challenges remain in the field of 3D cardiac tissue engineering. Functional immaturity relative to adult human myocardium persists, with engineered tissues typically exhibiting fetal-like characteristics in calcium handling, metabolism, and force generation. Addressing this limitation requires continued refinement of biophysical stimulation regimens, metabolic manipulation, and long-term culture strategies. Additionally, standardization across laboratories remains challenging due to variability in iPSC lines, protocols, and assessment methods. Future developments will likely focus on increasing model complexity through incorporation of immune cells, neural elements, and multi-tissue interactions, as well as enhancing analytical capabilities with advanced functional measurements and computational modeling. As these technologies continue to evolve, 3D engineered cardiac tissues and organoids will play an increasingly central role in advancing our understanding and treatment of cardiovascular disease [48] [54] [49].
Patient-specific human induced pluripotent stem cells (hiPSCs) have revolutionized the framework of precision medicine by providing a scalable, patient-specific platform that enables mechanistic insights into disease pathophysiology and therapeutic development [56] [57]. Generated by reprogramming adult somatic cells from patients into a pluripotent state, hiPSCs can self-renew indefinitely and differentiate into virtually any cell type, including cardiomyocytes, hepatocytes, and neurons [57] [58]. This technology bypasses ethical concerns associated with embryonic stem cells and offers high immunocompatibility as cells can be derived from a patient's own tissues [58]. Within cardiovascular research—a field where cardiovascular diseases (CVDs) remain the leading cause of mortality worldwide—hiPSC-derived cardiomyocytes (hiPSC-CMs) enable the modeling of genetic and acquired cardiac diseases in a patient-specific context, facilitating drug discovery, toxicity screening, and the development of tailored therapeutic interventions [56] [26].
Preparation of Therapeutic hiPSCs
The production of therapeutic-grade hiPSCs is a critical first step in the personalized medicine pipeline. This process involves collecting patient somatic cells, reprogramming them to pluripotency, and rigorously validating the resulting cell lines.
Somatic Cell Source Selection
The choice of somatic cell source significantly influences reprogramming efficiency, safety, and differentiation potential of resulting hiPSCs.
Table 1: Comparison of Somatic Cell Sources for hiPSC Generation
| Cell Source | Origin | Reprogramming Efficiency | Key Considerations |
|---|---|---|---|
| Dermal Fibroblasts | Mesoderm | Moderate | Gold standard; reliable but requires invasive biopsy [57] [58] |
| Peripheral Blood Cells | Mesoderm | High | Minimally invasive; enables collection from large donors [58] |
| Keratinocytes | Ectoderm | High | Accessible via minimally invasive hair follicle sampling [57] |
| Urinary Cells | Endoderm | Moderate | Non-invasive collection; useful for specific patient populations [58] |
Reprogramming Techniques
Various reprogramming methods have been developed, each with distinct advantages and limitations for clinical translation.
Table 2: hiPSC Reprogramming Techniques and Characteristics
| Method | Vector Type | Genomic Integration | Safety Profile | Efficiency |
|---|---|---|---|---|
| Sendai Virus | Viral | Non-integrating | High | High [58] |
| Episomal Plasmids | Non-viral | Non-integrating | High | Moderate [58] |
| mRNA | Non-viral | Non-integrating | High | Moderate [57] [58] |
| Retrovirus | Viral | Integrating | Low (oncogenic risk) | High [57] |
| Lentivirus | Viral | Integrating (excisable) | Moderate | High [57] [58] |
The original reprogramming method utilized retroviral vectors to deliver four transcription factors (OCT3/4, SOX2, c-MYC, KLF4), but this approach carries significant tumorigenic risk due to genomic integration and potential reactivation of oncogenes like c-MYC [57]. Current best practices favor non-integrating methods such as Sendai virus or episomal plasmids, which provide improved safety profiles while maintaining reasonable efficiency [58]. Recent advances include microRNA-based reprogramming (e.g., miR-302/367 cluster), which proceeds faster than traditional factor-based methods and avoids exogenous transcription factors [57].
Engineering Strategies for hiPSC-Based Therapeutics
Engineering approaches enhance hiPSC functionality and safety for therapeutic applications through biomodulation, genetic correction, and tissue engineering.
Genetic Engineering for Disease Correction
CRISPR-Cas9 genome editing enables precise correction of disease-causing mutations in patient-derived hiPSCs or introduction of specific mutations for disease modeling [56] [26]. This approach is particularly valuable for monogenic cardiovascular disorders such as familial hypertrophic cardiomyopathy or long QT syndrome, where isogenic pairs of corrected and uncorrected hiPSC lines can be generated to study disease mechanisms and validate therapeutic interventions [56].
Biomaterial-Enhanced Differentiation and Maturation
A significant limitation of hiPSC-CMs is their immature, fetal-like phenotype which limits their ability to model adult cardiac pathophysiology [56] [26]. Advanced biomaterials address this challenge by mimicking the native cardiac microenvironment:
- Hydrogels: Tunable mechanical properties and tissue-like elasticity enable replication of cardiac extracellular matrix (ECM) mechanics [56] [26]. Natural (e.g., collagen, fibrin) and synthetic hydrogels can be functionalized with adhesive peptides and growth factors to guide cellular organization.
- 3D Engineered Tissues: Combining hiPSC-CMs with biomaterials to create 3D tissue constructs that better recapitulate organ-level structure and function [56]. These systems demonstrate more mature electrophysiology, calcium handling, and contractile function compared to 2D cultures.
- Mechanical Conditioning: Application of physiological mechanical stimuli through stretch platforms or electrical pacing to promote structural and functional maturation [56].
Figure 1: hiPSC Cardiac Differentiation and Maturation Pathway: Key stages and engineering strategies for generating mature cardiomyocytes from hiPSCs.
Cardiovascular Disease Modeling with hiPSC-CMs
The application of hiPSC technology in cardiovascular disease modeling has provided unprecedented opportunities to study patient-specific disease mechanisms and screen therapeutic compounds.
Cardiac Mechanobiology in Health and Disease
The heart relies heavily on mechanical cues, with integrin- and cadherin-based adhesion complexes mediating mechanosensitive signaling that drives both physiological adaptation and pathological remodeling [56] [26].
Physiological Mechanotransduction: Cardiomyocytes sense mechanical cues through costameres, which align with Z-disks and contain integrins that connect the cytoskeleton to ECM proteins like collagen, fibronectin, and laminin [56]. Key mechanosensitive pathways include:
- Integrin-FAK-ERK: Force-sensitive proteins (talin, paxillin, vinculin) recruit intracellular mediators including focal adhesion kinase (FAK), initiating cascades such as Raf-MEK-ERK that drive cardiomyocyte hypertrophy and maturation [56].
- WNT Signaling: N-cadherin at intercalated discs binds and stabilizes β-catenin, a crucial component of adherens junctions involved in WNT signaling, which is targeted in standardized hiPSC-CM differentiation protocols [56] [26].
Pathological Remodeling: Under disease conditions, altered mechanical loading disrupts normal mechanotransduction, leading to maladaptive responses [56]. Studies using hiPSC-CMs have revealed that:
- Cytoskeletal proteins like filamin C (FLNC) maintain mechanical linkages, and truncation variants disrupt cytoskeletal stiffness, impair cell-ECM adhesion, and induce arrhythmic beating [26].
- Changes in ECM composition after cardiac injury cause reversion to fetal integrin expression patterns (upregulation of α5, α7, and β1D), promoting pathological remodeling [26].
Figure 2: Cardiac Mechanosensing Pathway: Mechanical forces are transduced into biochemical signals through integrin-mediated pathways.
Disease-Specific Modeling Applications
hiPSC-CMs have been successfully used to model various inherited and acquired cardiovascular disorders:
- Inherited Cardiomyopathies: Modeling hypertrophic cardiomyopathy (HCM) caused by mutations in sarcomeric proteins, where hiPSC-CMs recapitulate key features like hypertrophy, myofilament disarray, and arrhythmogenicity [56].
- Channelopathies: Investigation of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using patient-specific hiPSC-CMs, enabling drug testing and personalized therapeutic selection [56].
- Complex Acquired Diseases: Modeling diabetic cardiomyopathy, drug-induced cardiotoxicity, and mechanical overload conditions using hiPSC-CMs in combination with environmental stressors [56] [26].
Experimental Workflows and Methodologies
Robust experimental protocols are essential for generating reproducible, high-quality data from hiPSC-based disease models.
hiPSC-CM Differentiation Protocol
Standardized cardiac differentiation protocols primarily target the WNT signaling pathway to direct mesodermal and cardiac lineage specification [56] [26]:
- Mesoderm Induction (Days 0-2): Treat hiPSCs with GSK-3β inhibitor (CHIR99021) to activate WNT signaling, promoting commitment to mesodermal lineage expressing T Brachyury and TBX6 [56] [26].
- Cardiac Progenitor Specification (Days 2-5): Add WNT inhibitor (IWP-2 or IWR-1) to suppress WNT signaling, enabling cardiac mesoderm formation and expression of cardiac transcription factors (NKX2-5, TBX5, GATA4) [56].
- Cardiomyocyte Maturation (Days 5-30+): Culture in basal media with fatty acids, thyroid hormone (T3), and corticosteroids to promote metabolic and functional maturation [56]. Extended culture (≥80 days) with appropriate mechanical and electrical stimulation enhances structural and electrophysiological maturity [56] [26].
Functional Characterization Assays
Comprehensive assessment of hiPSC-CM function requires multiple complementary approaches:
- Contractile Function: Video-based analysis of contraction dynamics, force measurements using muscular thin films or traction force microscopy [56].
- Electrophysiology: Multi-electrode array (MEA) recordings for field potential measurements, patch clamping for action potential characterization [56] [26].
- Calcium Handling: Fluorescence imaging with calcium indicators (e.g., Fluo-4) to assess calcium transient dynamics and sarcoplasmic reticulum function [56].
- Structural Analysis: Immunostaining for sarcomeric organization (α-actinin, troponin), gap junctions (connexin-43), and mitochondrial network [56] [26].
Table 3: Essential Research Reagents for hiPSC-CM Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Reprogramming Factors | OCT4, SOX2, KLF4, c-MYC (Yamanaka factors) | Reprogram somatic cells to pluripotent state [57] [58] |
| Cardiac Differentiation Agents | CHIR99021 (GSK-3β inhibitor), IWP-2 (WNT inhibitor) | Direct cardiac lineage specification via WNT pathway modulation [56] [26] |
| Maturation Promoters | Thyroid hormone (T3), fatty acids, corticosteroids | Enhance structural and metabolic maturity of hiPSC-CMs [56] |
| Biomaterials | Fibrin, collagen, synthetic PEG hydrogels | Provide tissue-like 3D microenvironment for culture [56] [26] |
| Lineage Markers | Anti-cTnT, α-actinin, NKX2-5 antibodies | Identify and purify cardiomyocyte populations [56] |
Future Perspectives and Challenges
While hiPSC technology holds tremendous promise for precision medicine, several challenges must be addressed to realize its full potential.
Current limitations include the immature phenotype of hiPSC-CMs, heterogeneity in differentiation outcomes, and scalability for high-throughput applications [56] [26]. Future directions focus on enhancing maturation through advanced engineering approaches, including more physiologically relevant 3D culture systems, mechanical loading platforms, and multicellular integration to replicate native tissue complexity [56] [58]. Additionally, standardization of differentiation protocols and functional assessment methods across laboratories will be crucial for generating comparable data and advancing the field [56].
The integration of hiPSC technology with other emerging fields—including artificial intelligence for phenotypic analysis, organ-on-a-chip systems for multi-tissue interactions, and gene editing for precise disease modeling—will further expand its applications in personalized cardiovascular medicine [56] [58]. As these technologies mature, patient-specific hiPSC-based approaches are poised to transform drug discovery, disease modeling, and ultimately, clinical care for cardiovascular diseases.
Overcoming Challenges: hiPSC-CM Immaturity and Scalability
The application of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) in cardiovascular disease modeling and drug development represents a transformative advancement in personalized medicine. These cells, which retain the donor's genetic information, enable the study of disease mechanisms and therapeutic responses in a patient-specific context [59] [60]. However, a significant translational challenge persists: hiPSC-CMs consistently exhibit a fetal-like phenotypic state rather than maturing into adult-like cardiomyocytes [61] [20] [62]. This cellular immaturity manifests across three critical domains: structural, metabolic, and electrophysiological properties. The structural and functional gaps limit the fidelity of disease modeling, accuracy of drug screening, and potential for regenerative therapies [20] [60]. This technical review delineates these specific maturity gaps and synthesizes current experimental strategies being developed to bridge them, thereby enhancing the utility of hiPSC-CMs in cardiovascular research.
Structural and Functional Deficits in Immature hiPSC-CMs
Immature hiPSC-CMs display profound structural differences compared to adult cardiomyocytes, which directly impact their contractile function and predictive validity for adult cardiac physiology.
Morphological and Ultrastructural Characteristics
The table below summarizes the key structural differences between immature hiPSC-CMs and adult CMs.
Table 1: Structural and Functional Characteristics of Immature vs. Mature Cardiomyocytes
| Characteristic | Immature hiPSC-CMs / Fetal CMs | Mature Adult Cardiomyocytes |
|---|---|---|
| Cell Morphology | Round, small area (~30 μm²) [62] | Rod-shaped, anisotropic, large area (~150 μm²) [62] |
| Sarcomere Organization | Disorganized, rudimentary myofibrils [63] [20] | Highly organized, aligned myofibrils with distinct A-, I-, and Z-bands [63] |
| T-Tubules | Absent [20] | Elaborated network critical for excitation-contraction coupling [20] |
| Nucleation | Predominantly mononucleated [62] | Often binucleated or multinucleated [63] [62] |
| Sarcoplasmic Reticulum | Underdeveloped [60] | Well-developed, regulates Ca²⁺ release [60] |
Functional Consequences and Maturation Strategies
The disorganized sarcomeres and lack of T-tubules in hiPSC-CMs lead to inefficient force generation and impaired calcium-induced calcium release, resulting in weaker and slower contractions [63] [60]. Research demonstrates that prolonged culture (80-120 days) can drive significant structural maturation. Late-stage hiPSC-CMs show a doubling in cell size, increased anisotropy, greater myofibril density and alignment, and a ten-fold increase in multinucleation [63].
A primary strategy to promote structural maturity involves engineering a biomimetic microenvironment. Using nanofibrous scaffolds made of polymers like polycaprolactone (PCL) or polyurethane (PU) mimics the mechanical and topographical cues of the native cardiac extracellular matrix (ECM) [62]. Cells cultured on these aligned nanofibers develop a more elongated, anisotropic morphology and show upregulation of mature cardiac structural markers like MYH7 (β-myosin heavy chain) and TNNI3 (cardiac Troponin I) [62]. Other effective techniques include applying biophysical stimuli, such as mechanical stretching and electrical pacing, to drive the self-organization and alignment of contractile units [64] [20].
Metabolic Immaturity and the Fetal Energetic Profile
A defining feature of hiPSC-CM immaturity is their metabolic profile, which remains stuck in a fetal state and fails to execute the critical "metabolic switch" that occurs after birth.
The Fetal vs. Adult Metabolic Phenotype
Fetal cardiomyocytes and immature hiPSC-CMs rely primarily on glycolysis for ATP production, even in the presence of oxygen—a phenomenon known as the Warburg effect [61]. In contrast, adult cardiomyocytes are metabolic powerhouses that meet their high energy demands by preferentially oxidizing free fatty acids (FFAs) via mitochondrial oxidative phosphorylation, which generates significantly more ATP per molecule of substrate [61]. This metabolic transition is orchestrated by profound changes in mitochondria, which evolve from a small, round morphology with sparse cristae to a large, elongated form with dense, well-organized cristae that are tightly packed between sarcomeres [61].
Diagram: The Metabolic Switch in Cardiomyocyte Maturation
Experimental Protocols for Metabolic Maturation
Promoting metabolic maturation is crucial for generating hiPSC-CMs capable of sustaining adult-like contractile work. Key culture medium manipulations include:
- Substrate Shifting: Culturing hiPSC-CMs in media that is rich in free fatty acids (e.g., palmitate) and contains reduced glucose and no insulin, which forces a switch towards fatty acid β-oxidation [61].
- Hormonal Supplementation: Adding thyroid hormone (T3/T4) is a potent inducer of mitochondrial biogenesis and the expression of fatty acid oxidation enzymes [61].
- 3D Culture and Bioreactors: Growing hiPSC-CMs in 3D engineered heart tissues (EHTs) or using microfluidic systems enhances oxygen consumption rates and promotes mitochondrial oxidative capacity more effectively than 2D monolayers [61] [20].
Electrophysiological and Calcium Handling Deficiencies
The electrophysiological profile of hiPSC-CMs is a major determinant of their utility in modeling cardiac arrhythmias and predicting drug-induced cardiotoxicity, yet significant functional gaps remain.
Key Electrophysiological Limitations
The immature electrophysiological phenotype of hiPSC-CMs includes several critical deficiencies:
- Depolarized Resting Membrane Potential (RMP): A hallmark of immaturity is a depolarized RMP (around -50 to -60 mV) compared to the hyperpolarized RMP of adult ventricular CMs (around -80 to -90 mV) [63] [65]. This is largely due to low expression of the inward-rectifier potassium current (
IK1), which is essential for stabilizing the RMP in adult cells [65] [60]. - Spontaneous Automaticity: The low
IK1density, combined with the presence of pacemaker currents, causes hiPSC-CMs to beat spontaneously, unlike quiescent adult ventricular CMs [20] [60]. - Underdeveloped Calcium Handling: Immature hiPSC-CMs lack T-tubules and have a less developed sarcoplasmic reticulum (SR). Consequently, they rely on trans-sarcolemmal calcium influx rather than robust SR calcium-induced calcium release, leading to slower and less synchronous calcium transients [63] [60].
It is critical to note that technical artefacts can confound electrophysiological readings. Recent studies reveal that even with gigaohm seals, leak currents from patch-clamp pipettes can artificially depolarize the recorded membrane potential and distort action potential morphology, potentially exaggerating the observed immaturity [65].
Strategies for Functional Maturation
Advanced culture techniques are required to drive electrophysiological maturation:
- Prolonged Culture and Electrical Stimulation: Long-term culture (over 80 days) leads to a hyperpolarization of the RMP, increased action potential amplitude, and faster upstroke velocity [63]. Applying electrical field stimulation at a physiologically relevant pacing frequency (e.g., 1-2 Hz) can suppress automaticity and promote the development of adult-like ion channel expression and calcium handling properties [64] [20].
- Dynamic Clamp Technique: To acutely correct the
IK1deficiency, researchers use the "dynamic clamp" method. This real-time computational approach injects a computer-simulatedIK1current into the hiPSC-CM during patch-clamp recording, which successfully stabilizes the RMP and suppresses spontaneous activity [65]. - 3D Co-culture Models: Incorporating non-myocyte cells, such as cardiac fibroblasts, into 3D engineered tissues provides more physiologically relevant cell-cell interactions and paracrine signaling that support electrophysiological maturation [60].
The Scientist's Toolkit: Key Research Reagents and Solutions
The following table catalogs essential reagents and materials used in hiPSC-CM maturation protocols, along with their critical functions in bridging the maturity gaps.
Table 2: Essential Research Reagents for hiPSC-CM Maturation Studies
| Reagent / Material | Function / Application | Key Outcome / Rationale |
|---|---|---|
| CHIR99021 (GSK3 inhibitor) | Small molecule for Wnt/β-catenin activation; initiates cardiac differentiation [62] | Drives mesoderm specification toward cardiac lineage; high differentiation efficiency (>90%) [62] |
| IWP-2/Wnt-C59 (Wnt inhibitor) | Small molecule inhibiting Wnt secretion; used after CHIR99021 [62] | Promotes cardiac mesoderm specification and cardiomyocyte differentiation [62] |
| Polycaprolactone (PCL) Nanofibers | Synthetic, biodegradable polymer for engineered scaffolds [62] | Provides structural cues for cell alignment and elongation; enhances sarcomere organization [62] |
| Polyurethane (PU) Nanofibers | Synthetic polymer with tunable elasticity for scaffolds [62] | Mimics mechanical properties of native heart ECM; promotes maturation of contractile apparatus [62] |
| Triiodothyronine (T3) | Thyroid hormone added to maturation medium [61] | Potent inducer of mitochondrial biogenesis and metabolic switch to oxidative metabolism [61] |
| Fatty Acids (e.g., Palmitate) | Metabolic substrate in culture medium [61] | Forces cells to utilize fatty acid oxidation, mimicking adult cardiac metabolism [61] |
| CRISPR/Cas9 System | Genome editing tool for generating isogenic control lines [60] | Creates genetically matched controls; essential for validating disease-specific phenotypes [60] |
Diagram: An Integrated Workflow for hiPSC-CM Maturation
Addressing the structural, metabolic, and electrophysiological immaturity of hiPSC-CMs is a central challenge in cardiovascular research. No single intervention is sufficient to achieve a fully adult-like phenotype; instead, an integrated approach combining prolonged culture, biomimetic 3D environments, metabolic conditioning, and physiologically relevant biophysical stimulation shows the most promise [64] [20] [62]. As these strategies evolve and converge with emerging technologies like genome editing and AI-driven analysis, they will significantly enhance the predictive power of hiPSC-CM models [64] [37]. Closing these cellular maturity gaps is paramount for realizing the full potential of hiPSC-based platforms in de-risking drug discovery, elucidating complex disease mechanisms, and ultimately, developing personalized regenerative therapies for cardiovascular disease.
The application of human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) has revolutionized cardiovascular research, offering unprecedented opportunities for disease modeling, drug screening, and regenerative therapies [20]. However, a significant challenge persists: iPSC-CMs typically exhibit an immature, fetal-like phenotype that limits their fidelity in recapitulating adult human heart physiology [20] [66]. This immaturity manifests through disorganized sarcomeres, absent T-tubules, spontaneous automaticity, predominant glycolytic metabolism, and inadequate calcium handling [20] [66]. Overcoming these limitations is crucial for advancing the predictive accuracy of cardiac disease models and therapeutic applications.
This technical guide synthesizes current methodologies for enhancing iPSC-CM maturation, focusing on three cornerstone strategies: three-dimensional (3D) culture systems, electrical pacing, and metabolic priming. These approaches collectively address the multifactorial nature of cardiac maturation by replicating critical aspects of the native myocardial microenvironment—structural complexity, electromechanical stimulation, and energy metabolic shifts [67] [68]. The integration of these strategies within the context of cardiovascular disease modeling research provides a framework for generating more physiologically relevant in vitro platforms.
Core Maturation Strategies
Three-Dimensional (3D) Culture Systems
Transitioning from two-dimensional (2D) monolayers to three-dimensional cultures represents a paradigm shift in cardiac tissue engineering. 3D systems significantly enhance structural and functional maturation by promoting improved cell-cell and cell-extracellular matrix (ECM) interactions, essential for adult cardiomyocyte physiology [68].
Table 1: Types of 3D Cardiac Constructs and Their Applications
| Construct Type | Key Characteristics | Primary Applications | Notable Advantages |
|---|---|---|---|
| Engineered Heart Tissues (EHTs) | Scaffold-based (e.g., collagen, fibrin hydrogel); strip format; often includes non-myocytes [69]. | Disease modeling (e.g., HCM, DCM), contractile force measurement, drug testing [20] [69]. | Enables direct measurement of contractile force; recapitulates tissue-level anisotropy. |
| Cardiac Organoids | Self-organizing, scaffold-free or Matrigel-embedded; multicellular; mimics embryonic heart development [70] [69]. | Studying early cardiogenesis, congenital diseases, tissue regeneration [70] [69]. | Recapitulates embryonic heart structure and multi-lineage cellular crosstalk. |
| Cardiac Microtissues/Spheroids | Spherical, scaffold-free aggregates; mono- or multi-cellular [70]. | High-throughput drug screening, toxicity testing, disease modeling [70]. | Simple generation; suitable for high-throughput platforms; long-term culture viability. |
Experimental Protocols for 3D Culture
Protocol A: Generation of Scaffold-Free Human Organotypic Cardiac Microtissues (hOCMTs) [70] This protocol generates scaffold-free, beating microtissues that can be cultured long-term (over 100 days).
- 2D Cardiac Differentiation: Differentiate iPSCs in a 2D monolayer using a standard Wnt modulation protocol (e.g., GSK3 inhibition followed by PORCN inhibition).
- Dissociation and Aggregation: On day 15 of differentiation, dissociate the monolayer into single cells. Seed cells into round-bottom ultra-low attachment plates in cardiac maintenance medium supplemented with a ROCK inhibitor.
- 3D Culture and Maturation: After 48 hours, remove the ROCK inhibitor to allow spontaneous contraction. Continue culture with regular medium changes. Microtissues show spontaneous beating and cellular self-organization for several months.
Protocol B: Suspension Bioreactor Differentiation for High-Yield iPSC-CM Generation [14] This scalable protocol produces high-purity iPSC-CMs with improved functional maturity and batch-to-batch consistency.
- Quality Control: Use quality-controlled master cell banks of iPSCs. Confirm high pluripotency marker (e.g., SSEA4 >70%) for successful differentiation.
- Embryoid Body (EB) Formation: Aggregate iPSCs in a stirred bioreactor system to form EBs.
- Cardiac Differentiation: Initiate differentiation by adding Wnt activator CHIR99021 when EB diameter reaches ~100 µm. After 24 hours, replace with fresh medium. Following a 24-hour gap, add Wnt inhibitor IWR-1 for 48 hours.
- Maintenance and Harvest: Continue culture in cardiac maintenance medium. This protocol yields ~1.2 million cells/mL with >90% TNNT2+ cardiomyocytes by day 15.
Electrical Pacing
Exogenous electrical stimulation is a powerful tool to drive the electrophysiological and structural maturation of iPSC-CMs in 3D constructs by mimicking the native heart's electrical activity [68]. It promotes sarcomere alignment, enhances calcium handling, and fosters a shift from spontaneous to paced contraction.
Table 2: Electrical Stimulation Parameters and Their Functional Impacts in 3D Cultures
| Parameter | Typical Range | Impact on Maturation | Key Findings |
|---|---|---|---|
| Electric Field | 65 - 200 mV/mm [71] [67] | Drives structural and functional maturation. | Significantly increased CM component, TNNT2 expression, and calcium transient capacity in ECTs [67]. |
| Frequency | 1 - 6 Hz [67] [68] | Mimics physiological heart rates; improves force-frequency relationship (FFR). | Achievement of a positive FFR, a hallmark of adult CMs, indicating advanced maturation [67]. |
| Waveform | Biphasic, charge-balanced pulses [71] | Prevents electrode damage and pH shifts. | Promotes cardiac differentiation and avoids adverse electrochemical reactions [71]. |
| Duration | Chronic (days to weeks) [68] | Allows for sustained adaptive changes in gene expression and structure. | Leads to appearance of T-tubules, organized sarcomeric bands, and improved conduction velocity [68]. |
Experimental Protocol for Electrical Stimulation
Protocol: Simultaneous Electro-Dynamic Stimulation of Engineered Cardiac Tissues (ECTs) [67] This protocol applies combined electrical and mechanical stimulation to enhance ECT maturation synergistically.
- ECT Fabrication: Co-culture human iPSC-derived cardiovascular cells with Collagen I and Matrigel in engineered tissue constructs for 2 weeks.
- Stimulation Setup: Subject ECTs to one of four conditions for a defined period (e.g., 1-2 weeks):
- No stimulation (control)
- Dynamic (mechanical) stimulation alone
- Electrical stimulation alone (e.g., 65 mV/mm, 1 Hz)
- Simultaneous electro-dynamic stimulation
- Functional Assessment:
- Contractility: Measure force generation and force-frequency relationship.
- Calcium Handling: Assess calcium transient properties using fluorescent indicators.
- Histology: Evaluate structural maturation (sarcomere alignment, TNNT2 expression) and vascular network formation.
Metabolic Priming
The immature iPSC-CM relies primarily on glycolysis for energy production, similar to fetal cardiomyocytes. Mature adult cardiomyocytes, in contrast, derive over 70% of their ATP from mitochondrial oxidative phosphorylation, predominantly using fatty acids [66]. Directing the metabolic state of iPSC-CMs is therefore a critical lever for promoting maturation.
Experimental Protocol for Metabolic Maturation
Protocol: Maturation Media (MM) Formulation and Application [66] This protocol uses a tailored culture medium to induce a shift from glycolytic to oxidative metabolism.
- Baseline Differentiation: Differentiate iPSCs into cardiomyocytes using a standard protocol (e.g., in RPMI/B27 medium) until day 20.
- Maturation Media Formulation: Prepare MM by modifying the base medium to include:
- Lowered glucose (e.g., 3 mM initial) to discourage glycolysis.
- Supplementation with oxidative substrates: Albumin-bound fatty acids (AlbuMAX), creatine, L-carnitine, and taurine.
- Increased calcium to physiological levels suited for contractility.
- Application and Culture: At day 20, randomize iPSC-CM cultures to either conventional RPMI/B27 or MM. Maintain cultures for 3-5 weeks, changing media every 4 days. Monitor glucose depletion, which occurs rapidly in MM, forcing cells to utilize oxidative substrates.
Integrated Workflow and the Scientist's Toolkit
For successful implementation of maturation strategies, a structured workflow and the right tools are essential. The following diagram integrates the core strategies, and the subsequent table lists key reagents.
Table 3: The Scientist's Toolkit - Essential Research Reagents and Materials
| Category | Item | Function | Example Use |
|---|---|---|---|
| Small Molecules | CHIR99021 (GSK3 inhibitor) | Activates Wnt signaling to induce mesoderm and cardiac specification [14]. | Used in initial stages of both monolayer and bioreactor differentiation protocols. |
| IWR-1 (Wnt inhibitor) | Inhibits Wnt signaling to promote cardiac mesoderm differentiation into cardiomyocytes [14]. | Added after CHIR99021 to complete cardiac differentiation. | |
| Media Supplements | AlbuMAX / Fatty Acids | Provides essential lipids and promotes a shift from glycolysis to fatty acid oxidation [66]. | Key component of metabolic maturation media. |
| L-Carnitine, Creatine, Taurine | Supports mitochondrial fatty acid uptake and high-energy phosphate metabolism in mature CMs [66]. | Supplementation in maturation media to enhance bioenergetic capacity. | |
| Matrices & Scaffolds | Collagen I / Fibrin | Natural hydrogel scaffolds that provide a 3D microenvironment for cell growth and tissue formation [67]. | Base hydrogel for forming Engineered Heart Tissues (EHTs). |
| Matrigel | Complex basement membrane extract providing adhesion proteins and growth factors. | Often used mixed with collagen to support ECT formation and vascularization [67]. | |
| Specialized Equipment | Bioreactor / Spinner Flask | Provides controlled suspension culture (temperature, O₂, CO₂, pH, stirring) for scalable, consistent differentiation [14]. | Used for high-yield production of iPSC-CMs in 3D aggregates. |
| Electrical Stimulator | Applies controlled, rhythmic electrical pulses to engineered tissues to promote electrophysiological maturation [71] [67]. | Used for chronic pacing of ECTs to induce adult-like properties. |
Application in Cardiovascular Disease Modeling
Maturation strategies are not merely about achieving an adult phenotype; they are critical for creating clinically predictive models of cardiovascular disease. Immature iPSC-CMs often fail to recapitulate key pathological hallmarks, leading to false negatives or positives in drug screening and disease modeling.
Enhanced Disease Phenotype Recapitulation: Mature iPSC-CMs exhibit greater reliance on sodium channels for depolarization and improved sarcoplasmic reticulum calcium cycling. This is crucial for accurately modeling diseases like Long QT Syndrome Type 3 (LQT3), which is linked to mutations in the cardiac sodium channel gene SCN5A. In immature CMs, which are resistant to sodium channel blockers, LQT3 phenotypes may be muted. Metabolic maturation makes the action potential dependent on sodium current, allowing for robust modeling of LQT3 and reliable testing of sodium channel-blocking drugs [66]. Similarly, mature models are essential for capturing the contractile deficits in Dilated Cardiomyopathy (DCM) linked to mutations in genes like RBM20, which require robust calcium cycling and force generation to manifest [66].
Drug Screening and Toxicity Testing: Mature tissues provide a more accurate platform for assessing drug efficacy and cardiotoxicity. The presence of a positive force-frequency relationship (FFR)—a characteristic of adult myocardium that is absent in immature tissue—is critical for evaluating inotropic drugs [67]. Furthermore, the enhanced structural organization and metabolic state of matured ECTs lead to more predictive responses to chemotherapeutic agents known to cause cardiotoxicity (e.g., doxorubicin), as they better reflect the metabolic vulnerabilities of the adult heart [70].
The pursuit of mature iPSC-derived cardiomyocytes is a central challenge in cardiovascular research. The integration of 3D culture systems, electrical pacing, and metabolic priming addresses the multifaceted nature of cardiac development, driving iPSC-CMs toward an adult-like phenotype. These strategies work synergistically: 3D architecture provides the structural context, electrical stimulation imposes a functional workload and rhythm, and metabolic priming ensures the underlying energy substrate flexibility and capacity required for adult function.
As these protocols continue to be refined and standardized, their integration will undoubtedly enhance the fidelity of in vitro disease models, improve the predictive power of drug screening and toxicity assays, and ultimately accelerate the development of novel therapies for cardiovascular diseases. Future efforts should focus on standardizing these protocols across laboratories, further elongating culture periods, and integrating multiple cell types to fully mimic the native heart's cellular complexity and immune environment.
In the field of cardiovascular disease modeling research, induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) have emerged as a transformative platform for studying disease mechanisms and screening therapeutic compounds. However, the full potential of this technology is hampered by a persistent challenge: significant batch-to-batch and line-to-line variability in differentiation outcomes. This variability manifests as inconsistencies in differentiation efficiency, cellular purity, functional maturity, and pharmacological responses across different experimental repetitions and iPSC lines [14] [72]. For cardiovascular disease modeling and drug development professionals, this lack of reproducibility threatens the reliability of experimental data and complicates the interpretation of results. The inherent complexity of cardiac differentiation, where multiple signaling pathways must be precisely coordinated across temporal stages, creates numerous opportunities for variation to emerge [73]. This technical guide examines the principal sources of this variability and presents a comprehensive framework of evidence-based strategies to standardize differentiation protocols and enhance the reproducibility of iPSC-CM generation for cardiovascular research applications.
The journey from iPSCs to functional cardiomyocytes is susceptible to variability at multiple critical junctures. Understanding these sources is essential for implementing effective quality control measures.
Input Cell Quality and Pluripotency Status: The initial quality and state of iPSCs significantly influence differentiation efficiency. Variations in pluripotency marker expression (e.g., SSEA4) directly correlate with successful cardiac differentiation. Research has demonstrated that low SSEA4 expression (<70%) predetermines failed differentiations (<90% TNNT2+ cells), while high SSEA4 (>70%) correlates with efficient differentiations (>90% TNNT2+) [14]. Furthermore, the somatic cell source and reprogramming method used to generate iPSCs can create line-to-line variability, as different epigenetic memories may influence differentiation propensity [72] [38].
Protocol Sensitivity and Temporal Dynamics: Cardiac differentiation protocols typically involve precisely timed activation and inhibition of key signaling pathways, particularly Wnt/β-catenin. Minor deviations in the timing, duration, or concentration of differentiation factors can dramatically alter outcomes. For instance, in suspension culture systems, the size of embryoid bodies (EBs) at the time of Wnt activation critically determines efficiency. EBs smaller than 100μm may disassemble upon CHIR treatment, while those larger than 300μm differentiate less efficiently due to diffusion limitations [14]. Similarly, in monolayer differentiation, the optimal duration of CHIR incubation may vary between cell lines, requiring line-specific optimization [14].
Environmental and Handling Factors: The culture environment and handling techniques introduce another layer of variability. Temperature fluctuations during cryopreservation or cell passaging can trigger mitochondrial dysfunction and delayed cell death [74]. Local heterogeneity in cell seeding density in monolayer cultures creates microenvironments that affect differentiation efficiency, while static culture conditions can result in suboptimal nutrient distribution and pH buffering [14]. Additionally, the transition between culture platforms (2D vs. 3D) and the specific extracellular matrices used can further contribute to inconsistent differentiation outcomes [30] [75].
Process Engineering and Systematic Optimization Approaches
Implementing statistical and systematic approaches to process optimization represents a powerful strategy for enhancing reproducibility and controllability in cardiac differentiation.
Design of Experiments (DoE) for Controlled Co-differentiation: The application of statistical Design of Experiments (DoE) methodology enables researchers to efficiently optimize multiple process parameters simultaneously. This approach has been successfully implemented for cardiac multilineage co-differentiation processes, where researchers divided the process into two stages—cardiogenic mesoderm induction and subsequent trilineage co-differentiation—allowing for stage-specific optimization [73]. Using sequential DoE, researchers achieved approximately 95% induction efficiency of KDR+/PDGFR-α+ cardiogenic mesoderm cells from iPSCs with minimal batch-to-batch variability by carefully optimizing activin A and CHIR-99021 concentrations [73]. In the trilineage co-differentiation stage, unique multi-response models delineated differentiation ratios within a defined parameter space of WNT signal inhibitor and VEGF, enabling identification of conditions that steer co-differentiation toward desired cellular constitutions [73].
Stirred Suspension Bioreactor Systems: Transitioning from traditional monolayer cultures to controlled suspension systems addresses several sources of variability inherent in static cultures. Optimized stirred suspension bioreactor protocols have demonstrated remarkable consistency, producing ~1.21 million cells per mL with >90% TNNT2+ purity across 25 differentiations of 14 different iPSC lines [14]. These systems provide continuous monitoring and adjustment of temperature, O₂, CO₂, and pH, while constant mixing ensures homogeneous distribution of nutrients, differentiation factors, and cellular aggregates. The scalability of bioreactor systems (from 2.5 to 1000 mL) further enhances their utility for generating reproducible cell batches for high-throughput applications [14].
Table 1: Quantitative Comparison of Differentiation Platforms
| Parameter | Traditional Monolayer | Stirred Suspension Bioreactor |
|---|---|---|
| Average Yield | Variable across protocols | ~1.21 million cells/mL [14] |
| Purity (%TNNT2+) | ~85-95% (line-dependent) [72] | ~94% (consistent across lines) [14] |
| Inter-batch Variability | Higher | Significantly reduced [14] |
| Scalability | Limited by surface area | Highly scalable (linear volume increase) [14] |
| Onset of Contraction | Day 7 [14] | Day 5 [14] |
| Cryopreservation Recovery | Reduced viability and function [14] | >90% viability [14] |
Advanced Differentiation and Progenitor Cell Strategies
Novel approaches focusing on intermediate progenitor stages and alternative differentiation pathways offer promising avenues for enhancing reproducibility and maturation.
Progenitor Reseeding and Cryopreservation: A recently developed method to improve CM purity involves detaching and reseeding progenitors between the EOMES+ mesoderm and ISL1+/NKX2-5+ cardiac progenitor stages. This approach improves CM purity by 10-20% (absolute) without negatively affecting contractility, sarcomere structure, or CM number [75]. Crucially, researchers have demonstrated that these specific progenitor populations (EOMES+ mesoderm and ISL1+/NKX2-5+ cardiac progenitors) remain viable and functionally competent after cryopreservation, enabling the creation of master progenitor banks for on-demand CM production [75]. This strategy facilitates the transition to defined extracellular matrices, including fibronectin, vitronectin, and laminin-111, all of which supported continued differentiation of reseeded progenitors to CMs [75].
Alternative Differentiation Pathways Using Sfrp2: Replacing broad-spectrum Wnt pharmacological inhibitors with specific Wnt inhibitors represents another strategy to enhance maturation and reproducibility. The substitution of conventional inhibitors with Sfrp2, a specific Wnt3a inhibitor, has been shown to produce more mature cardiomyocytes as evidenced by improved sarcomere structure, reduced circularity, longer sarcomeres, lower beating frequency, and the formation of polarized gap junctions [76]. This specificity potentially reduces unintended off-target effects that contribute to variability. From a mechanistic perspective, Sfrp2 functions by inhibiting Wnt3a, thereby modulating the β-Catenin pathway at a specific developmental point corresponding to cardiac mesoderm specification [76].
Diagram 1: Progenitor Reseeding and Cryopreservation Workflow. This diagram illustrates the experimental workflow for improving cardiomyocyte differentiation purity through intermediate progenitor reseeding and cryopreservation, enabling the creation of progenitor banks for on-demand CM production [75].
Quality Control and Characterization Frameworks
Implementing rigorous quality control checkpoints throughout the differentiation process is essential for identifying and correcting variability before it compromises experimental outcomes.
Input Cell Quality Assessment: Establishing quality-controlled master cell banks with comprehensive characterization is a fundamental first step. Recommended quality control measures include karyotyping to ensure genetic stability, mycoplasma testing, and quantification of pluripotency markers (e.g., SSEA4 >70% via FACS) [14]. Monitoring EB diameter in suspension cultures (targeting 100μm at CHIR addition) provides a simple but critical morphological checkpoint, as significant deviations from this optimal size range dramatically impact differentiation efficiency [14].
Functional and Molecular Characterization of Output: Comprehensive characterization of differentiated CMs should extend beyond purity assessment to include functional and molecular maturity metrics. Key parameters include sarcomere structure and length (longer sarcomeres indicate greater maturity), electrophysiological profiling (action potential duration, beating frequency), multinucleation, junctional Cx43 expression, and subtype specification (ventricular vs. atrial) [76] [75]. Gene expression analysis of maturation markers (MYH7, MYL2, MYL3 for ventricular identity; ACTN2, TNNT2 for structural proteins) provides molecular validation of functional observations [14] [76].
Table 2: Key Quality Control Checkpoints and Target Metrics
| Checkpoint | Assessment Method | Target Metric | Impact on Differentiation |
|---|---|---|---|
| Input iPSCs | FACS for SSEA4 | >70% SSEA4+ [14] | Predetermines successful differentiation (>90% TNNT2+) |
| EB Formation | Diameter measurement | 100μm [14] | Optimized response to CHIR; avoids disintegration or diffusion limits |
| Mesoderm Induction | FACS for KDR+/PDGFR-α+ | ~95% efficiency [73] | High-quality progenitor population for trilineage differentiation |
| CM Purity | FACS for TNNT2 | >90% [14] | Sufficient yield for experiments and applications |
| CM Maturity | Sarcomere length | >1.6μm [76] | Enhanced structural and functional maturity for disease modeling |
| Functional Output | Beating frequency | Lower frequency indicates greater maturity [76] | More physiologically relevant responses in drug testing |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Standardized Cardiac Differentiation
| Reagent/Material | Function | Application Notes |
|---|---|---|
| CHIR99021 | GSK-3β inhibitor; activates Wnt signaling | Concentration and duration require optimization per cell line [14] [76] |
| Sfrp2 | Specific Wnt3a inhibitor; promotes maturation | Alternative to broad-spectrum Wnt inhibitors; enhances sarcomere structure and gap junction formation [76] |
| Y-27632 (ROCK inhibitor) | Enhances cell survival after passaging | Typically used for 24h post-passage; improves plating consistency [76] |
| Defined Matrices (Vitronectin, Laminin-111) | Provides substrate for cell attachment and signaling | Supports progenitor reseeding; more defined than biological extracts [75] |
| IWR-1 | Wnt pathway inhibitor; promotes cardiac specification | Used after CHIR-mediated activation; timing critical for efficiency [14] |
| B27 Supplement (minus insulin) | Defined culture supplement | Supports cardiac differentiation; insulin-free version used in early stages [73] |
Diagram 2: Integrated Quality Control Framework for Cardiac Differentiation. This diagram outlines the key decision points and monitoring stages in a standardized cardiac differentiation workflow, emphasizing critical process control points that require careful monitoring to minimize batch-to-batch variability [73] [14] [75].
The standardization of iPSC differentiation to cardiomyocytes represents a critical advancement for realizing the full potential of this technology in cardiovascular disease modeling and drug development. Through the implementation of systematic process engineering approaches, controlled culture platforms, intermediate progenitor strategies, and comprehensive quality control frameworks, researchers can significantly reduce batch-to-batch variability and enhance experimental reproducibility. The integration of these methodologies provides a solid foundation for generating high-quality, physiologically relevant iPSC-CMs that reliably model cardiovascular diseases and produce consistent results in compound screening. As these standardized approaches continue to evolve and gain widespread adoption, they will accelerate the translation of iPSC technology into clinically relevant applications and ultimately advance the field of personalized cardiology.
The application of induced pluripotent stem cells (iPSCs) in cardiovascular disease modeling and therapy development represents a paradigm shift in regenerative medicine. These cells offer an unprecedented opportunity to generate patient-specific cardiomyocytes for disease modeling, drug screening, and potentially cell replacement therapy for heart failure [77] [54]. However, translating this promise into clinically viable therapies necessitates overcoming two interconnected hurdles: ensuring the safety of iPSC-derived products by mitigating tumorigenicity risks and establishing robust, scalable Good Manufacturing Practice (GMP) processes. The tumorigenic risk stems primarily from residual undifferentiated pluripotent stem cells that may form teratomas or from the uncontrolled proliferation of partially reprogrammed cells [78] [79]. Simultaneously, manufacturing processes must be designed to consistently produce high-quality, well-characterized cell products at a scale suitable for clinical applications, while maintaining strict adherence to GMP standards [80]. This technical guide examines these challenges within the context of cardiovascular research, providing a framework for navigating the complex pathway from laboratory discovery to therapeutic application.
Tumorigenicity Risks in iPSC-Derived Cardiac Products
Origins and Mechanisms of Tumor Formation
The tumorigenic potential of iPSC-derived products constitutes the most significant safety concern for clinical translation. Multiple mechanisms contribute to this risk, beginning with the reprogramming process itself. The original Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC) include the proto-oncogene c-MYC, whose deregulated expression occurs in a wide range of human cancers [78]. Residual undifferentiated iPSCs persisting in differentiation cultures can form teratomas upon transplantation, as demonstrated in multiple pre-clinical studies. For instance, Kim et al. tracked engraftment of iPSCs in a SCID murine model of myocardial infarction and demonstrated teratoma formation despite reported functional improvement [77]. Beyond teratoma formation, genomic instability acquired during reprogramming presents additional concerns. iPSCs exhibit higher rates of chromosomal aberrations, copy number variations, and single nucleotide variants compared to embryonic stem cells, potentially creating oncogenic precursors [78].
The heterogeneity of cellular products derived from iPSCs further compounds these risks. Regardless of the reprogramming strategy, final cell preparations may contain a mixture of target cells, residual undifferentiated iPSCs, and partially differentiated cells with teratoma-forming potential [78]. The complex in vivo environment can then enable uncontrolled integration and proliferation of these cells. This risk is particularly concerning in cardiovascular applications where iPSC-derived cardiomyocytes are transplanted into already compromised myocardium, as the inflammatory and hypoxic environment might selectively promote the survival of undifferentiated cells [77].
Table 1: Tumorigenicity Risk Factors in iPSC-Derived Cardiovascular Therapies
| Risk Category | Specific Factors | Potential Consequences |
|---|---|---|
| Reprogramming Method | Use of integrating vectors (retrovirus, lentivirus), Proto-oncogenes (c-MYC), Incomplete vector silencing | Genomic instability, insertional mutagenesis, oncogene activation |
| Cell Product Composition | Residual undifferentiated iPSCs, Partially reprogrammed cells, Heterogeneous differentiation | Teratoma formation, uncontrolled proliferation of non-target cells |
| Genomic Integrity | Chromosomal aberrations, Copy number variations, Single nucleotide variants | Oncogenic transformation, abnormal growth behavior |
| Post-Transplantation Environment | Inflammatory milieu, Hypoxic conditions, Mechanical stress | Selective survival advantage for undifferentiated cells, Malignant transformation |
Strategies for Risk Mitigation
Optimizing Reprogramming and Differentiation
Significant progress has been made in developing strategies to minimize tumorigenic potential through improved reprogramming techniques. The replacement of c-MYC with alternative factors represents a primary approach. Studies demonstrate that L-Myc and N-Myc can reduce teratoma incidence in iPSCs while maintaining reprogramming efficiency [78]. Chemical induction represents another promising avenue, using small molecules to replace specific transcription factors. Valproic acid (a histone deacetylase inhibitor), RepSox, and CHIR99021 have successfully substituted for Sox2 and c-Myc in various protocols [78]. Pushp et al. argued that cocktails containing transcription factors and small molecules yield better reprogramming efficiency than chemical cocktails alone, while exhibiting less tumorigenicity than transcription factors only [78].
For cardiac differentiation, robust protocols that maximize efficiency and purity are essential. Research indicates that modulating key signaling pathways at specific timepoints guides successful cardiomyocyte differentiation. The Wnt/β-catenin signaling pathway requires precise temporal control—activation during early mesoderm induction followed by inhibition during cardiac specification [54]. Ren et al. demonstrated that pre-treatment with BMP-4 of human iPSCs, followed by post-treatment with a small molecule Wnt inhibitor, significantly boosts the formation of cardiomyocytes with normal electrophysiological function [54]. Additional maturation strategies including 3D culture, electrical pacing, extended culture, and fatty acid supplementation enhance the functional and structural maturity of iPSC-derived cardiomyocytes, potentially reducing their susceptibility to aberrant behavior [54].
Purification and Elimination Strategies
Effective removal of residual undifferentiated iPSCs from final products constitutes a critical safety step. Multiple approaches have been developed targeting pluripotent cell-specific markers. These include antibody-based magnetic cell sorting, fluorescence-activated cell sorting, and chemical inhibition [79]. Additional innovative approaches leverage the differential metabolic requirements of pluripotent versus differentiated cells. For example, providing lactate as an energy source enriches for cardiomyocytes, which can efficiently utilize lactate, while undifferentiated iPSCs are selectively eliminated [54].
Genetic strategies employing "suicide genes" or drug-inducible caspase systems provide an additional safety layer by enabling selective elimination of proliferating undifferentiated cells if necessary after transplantation [78]. These systems can be designed to activate in response to specific signals or administered drugs, providing a controllable safety mechanism.
Table 2: Methods for Tumorigenic Risk Reduction in iPSC-Derived Therapies
| Strategy Category | Specific Methods | Key Advantages | Limitations |
|---|---|---|---|
| Reprogramming Optimization | Myc-free protocols (L-Myc), Chemical induction, Non-integrating vectors | Reduced oncogenic potential, Lower genomic instability | Often lower efficiency, More complex protocols |
| Cell Purification | FACS/MACS using pluripotency markers, Metabolic selection (lactate), MicroRNA switches | High specificity, Scalable, Compatible with GMP | Potential cell loss, Marker variability |
| Genetic Safety Switches | Drug-inducible caspase systems, Suicide genes (HSV-TK) | Controllable elimination, Post-transplantation safety | Additional genetic modification, Immunogenicity risk |
| Process Control | Automated monitoring, CPP/CQA implementation, In-process analytics | Consistent product quality, Early risk detection | Requires significant validation, Infrastructure investment |
GMP Manufacturing and Scale-Up Considerations
Establishing Robust Manufacturing Processes
The transition from research-scale to clinically applicable manufacturing requires meticulous process development and control. A typical iPSC therapy development pipeline involves multiple critical steps: isolation of primary cells, reprogramming to induce pluripotency, clone selection, master and working cell bank establishment, directed differentiation, and final product formulation [80]. At each stage, maintaining pluripotency, genetic stability, and differentiation potential must be carefully balanced with scalability and reproducibility requirements.
A fundamental consideration in GMP manufacturing is the move from feeder-dependent to feeder-free culture systems. While feeder layers (mouse or human fibroblasts) provide essential growth factors and extracellular matrix components that support pluripotent cell growth, they introduce significant challenges for scaled, regulated manufacturing [81]. Feeder-based systems are labor intensive, hard to scale, and pose risks of animal pathogen and mycoplasma contamination [81]. Furthermore, feeder cells can interfere with molecular and flow cytometry experiments during quality control testing [81]. Multiple matrices have been successfully employed for feeder-free culture, including Geltrex, Matrigel, and recombinant laminin-521, combined with specialized media such as mTeSR or StemFlex [81].
Process control relies heavily on defining and monitoring Critical Process Parameters (CPPs) and Critical Quality Attributes (CQAs) throughout manufacturing. CPPs include factors such as cell seeding density, passage method, dissociation reagents, and media composition changes, all of which significantly impact final product quality [80]. CQAs encompass measures of cell viability, identity, purity, potency, and safety, requiring robust analytical methods for assessment.
Analytical Technologies and Process Monitoring
Advanced analytical technologies play a crucial role in ensuring product quality and process consistency throughout manufacturing. Sartorius offers integrated solutions including the Incucyte and iQue platforms, which provide high-capacity characterization of cell phenotype and function with unprecedented speed and depth [80]. These systems enable easy tracking of changing phenotypes during differentiation and rapid sampling with low volume requirements.
For clone selection, the CellCelector system offers the ability to select iPSC clones or areas of clones of interest for further expansion in a fast and gentle fashion [80]. This technology bridges early-stage development with manufacturing, ensuring selection of optimal founder populations.
Recent advancements in artificial intelligence and machine learning offer transformative potential for manufacturing control. Building an expert system for quantitative monitoring of iPSCs during expansion, researchers have developed an imaging and image analysis pipeline consisting of a rapid imaging system, a 2D U-Net for segmentation, a 3D U-Net for mitosis detection, and a tracking algorithm that allows tens of thousands of individual iPSCs and their progeny to be segmented and tracked over time and multiple cell divisions [82]. This approach enables the quantification of characteristics including morphology, migration rate, mitosis rate, time between mitoses, and expression of fluorophores associated with transcription factors, providing unprecedented insight into population dynamics and heterogeneity.
Automation represents another critical component of scalable GMP manufacturing. Automated systems reduce operator-to-operator variability, facilitate higher throughput, and minimize the risk of contamination [80]. As noted by Nicola Bevan of Sartorius, increasing the amount of process automation reduces the risk of cell infections and minimizes hands-on time in the lab [80].
The Scientist's Toolkit: Essential Reagents and Technologies
Table 3: Key Research Reagent Solutions for iPSC Therapy Development
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Reprogramming Factors | OCT4, SOX2, KLF4, L-MYC (OSKL), Small molecules (VPA, CHIR99021, RepSox) | Induce pluripotency in somatic cells | L-Myc reduces tumorigenicity vs. c-MYC; Small molecules improve safety profile |
| Culture Matrices | Geltrex, Matrigel, Laminin-521 | Provide extracellular matrix support for feeder-free culture | Laminin-521 shows excellent attachment and survival; Defined matrices preferred for GMP |
| Culture Media | mTeSR, StemFlex, KO-DMEM with KSR | Support pluripotent stem cell growth and maintenance | StemFlex enhances cloning efficiency; Formulations increasingly xeno-free |
| Differentiation Factors | BMP4, Activin A, CHIR99021, IWR-1, VEGF, FGF2 | Direct lineage-specific differentiation | Temporal control crucial; Small molecules improve reproducibility and cost |
| Cell Dissociation | Gentle Cell Dissociation Reagent, TrypLE, Accutase | Passage cells while maintaining viability | Gentle enzymes improve recovery; Reduce spontaneous differentiation |
| Cryopreservation | CRYOSTEM, DMSO-based media, RevitaCell | Maintain cell viability during freeze-thaw | Controlled-rate freezing critical; RevitaCell improves post-thaw recovery |
Integrated Workflows and Future Directions
The successful clinical translation of iPSC-based cardiovascular therapies requires seamless integration of safety and manufacturing considerations from the earliest stages of development. This begins with careful selection of starting cell sources, continues through reprogramming and banking strategies, and extends to differentiation, purification, and final product formulation. Emerging technologies such as chemical reprogramming, which offers a potentially safer alternative to genetic methods, and point-of-care manufacturing systems that automate much of the production process, represent promising avenues for future development [78] [83].
Quality control must encompass multiple orthogonal methods to thoroughly assess product safety. Recommended assessments include genomic integrity evaluation (karyotyping, CNV analysis), pluripotency marker detection (flow cytometry, immunocytochemistry), functional potency assays, and sterility testing [79] [80]. The Microsart ATMP Sterile Release Kit, for example, is specifically designed to meet the need for fast and accurate sterility testing of Advanced Therapy Medicinal Products with short shelf-lives [80].
The following diagram illustrates an integrated workflow for developing and manufacturing iPSC-derived cardiovascular therapies with built-in safety controls:
Integrated Workflow for iPSC-Derived Cardiovascular Therapies
Looking ahead, the field continues to evolve with several promising developments. The application of artificial intelligence and machine learning for predictive modeling of differentiation outcomes and product quality shows significant potential [82]. Similarly, the development of increasingly sophisticated metabolic maturation strategies to generate iPSC-derived cardiomyocytes that more closely resemble adult rather than fetal cells will enhance both the safety and efficacy of resulting therapies [54]. As these technologies mature, they will further enable the robust, scalable manufacturing of safe and effective iPSC-based therapies for cardiovascular disease.
The following diagram illustrates the critical signaling pathway involved in the efficient differentiation of iPSCs to cardiomyocytes, which is essential for generating pure populations of target cells:
Cardiac Differentiation Signaling Pathway
The field of cardiovascular disease (CVD) modeling is undergoing a profound transformation, driven by the synergistic integration of induced pluripotent stem cell (iPSC) technology, CRISPR-based gene editing, and artificial intelligence (AI). This convergence is establishing a new paradigm for quality control (QC) in preclinical research, enabling unprecedented precision in modeling genetic cardiovascular disorders and accelerating the drug discovery pipeline. iPSCs provide a unique human-relevant platform for disease modeling, but their full potential is constrained by inherent variability and the genetic heterogeneity of patient-derived lines. The combination of CRISPR gene editing and AI-powered analytics directly addresses these limitations by establishing robust, standardized, and isogenic model systems with enhanced predictive validity. This technical guide examines the integral role of these technologies in creating a next-generation QC framework for iPSC-based cardiovascular research, detailing specific methodologies, applications, and standardized protocols for the research community.
Foundational Technologies and Their Synergies
iPSC-Derived Cardiomyocytes in Cardiovascular Research
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) offer a patient-specific, ethically sound, and scalable platform for modeling cardiovascular diseases in vitro. However, a significant challenge in their application is their characteristic immaturity, which manifests in morphological, electrophysiological, and metabolic differences compared to adult human cardiomyocytes (AdCMs). As detailed in [3], hiPSC-CMs are typically smaller and more rounded, possess disorganized sarcomeres, lack developed T-tubule systems, and exhibit a primarily glycolytic metabolism, unlike the fatty acid-oxidizing AdCMs. This immature state can limit the accuracy of disease phenotyping and drug response predictions, making quality control and maturation critical areas of focus.
CRISPR Gene Editing for Precision Disease Modeling
CRISPR-Cas9 technology enables precise genetic modifications in iPSCs, which is fundamental for establishing high-quality, controlled disease models. Its primary application in QC is the creation of isogenic control lines. These are pairs of iPSC lines—typically a disease-carrying line and its genetically corrected counterpart, or a healthy line into which a disease-causing mutation has been introduced—that share an identical genetic background [84]. This strategy isolates the phenotypic effects of a specific mutation from the confounding noise of broader genetic variability, thereby greatly enhancing the reliability of disease mechanism studies and drug screening outcomes [84]. Advanced CRISPR systems, including base editors and prime editors, further improve precision and reduce off-target effects, contributing to higher-quality genetic models [85].
Artificial Intelligence and Machine Learning in QC
AI and machine learning (ML) are being deployed across the iPSC workflow to automate, standardize, and enhance QC processes. The U.S. Food and Drug Administration (FDA) has recognized the surge in drug applications incorporating AI/ML components and is developing a risk-based regulatory framework to support their use [86]. In the context of iPSC and CRISPR workflows, AI applications include:
- Predictive Modeling: Using AI algorithms to predict optimal gRNA designs for CRISPR editing to maximize on-target efficiency and minimize off-target effects [84].
- Phenotypic Analysis: Employing high-content imaging data from iPSC-CMs to train ML models for automated classification of cell maturity, sarcomere organization, and disease-specific phenotypes [87].
- Data Integration: AI systems can integrate multi-omics data (genomic, transcriptomic, proteomic) from isogenic iPSC-CMs to identify novel biomarkers of disease and treatment response, generating more nuanced QC metrics [88].
Table 1: Core Technologies and Their Quality Control Functions
| Technology | Primary QC Function | Key Advantage for CVD Modeling |
|---|---|---|
| iPSC Platforms | Provides human-relevant cellular substrate | Recapitulates patient-specific genetics and pathophysiology [3] |
| CRISPR-Cas9 | Generates isogenic controls | Controls for genetic background, isolating mutation-specific effects [84] |
| AI/ML Analytics | Automates phenotypic analysis & predicts editing outcomes | Provides objective, high-throughput QC; enhances editing precision [84] [87] |
Quality Control Applications and Experimental Workflows
Establishing Isogenic iPSC Lines for CVD Modeling
The generation of isogenic iPSC lines is a cornerstone of quality control in genetic disease modeling. The workflow below outlines the key steps, from design to validation, for creating a hypertrophic cardiomyopathy (HCM) model using a common mutation in the MYBPC3 gene.
Diagram 1: Creating an isogenic control line.
Detailed Protocol: Generating an Isogenic Control for HCM
- gRNA and Donor Template Design: Design a single-guide RNA (sgRNA) targeting the genomic region near the MYBPC3 mutation (e.g., c.1504C>T). Co-design a single-stranded oligodeoxynucleotide (ssODN) donor template containing the wild-type sequence with the corrective base change, flanked by homologous arms (~60-90 bp each) [84].
- CRISPR-Cas9 Delivery: Deliver the Cas9 ribonucleoprotein (RNP) complex (consisting of Cas9 protein and sgRNA) along with the ssODN donor into patient-derived iPSCs using electroporation. Using RNP complexes, as opposed to plasmid vectors, reduces the time of exposure and potential for off-target editing [89].
- Single-Cell Cloning: After transfection, plate cells at a very low density to allow for the isolation of single-cell-derived clones. Manually pick and expand individual colonies [84].
- Genotypic Validation: Screen expanded clones by Sanger sequencing across the target locus to identify those with the successful correction. Confirm the absence of unintended modifications at potential off-target sites (predicted by in silico tools) using next-generation sequencing (NGS) [84] [85].
- Phenotypic Validation: Differentiate the genetically corrected clone and the original mutant iPSC line into cardiomyocytes. Assess functional rescue by comparing key phenotypic readouts, including calcium transient imaging (to assess Ca²⁺ handling) and contractility analysis using video-based edge detection, to confirm that the correction has normalized the disease phenotype [3].
AI-Enhanced Functional Phenotyping and Maturation
A major QC challenge is the functional assessment of hiPSC-CMs, which is traditionally low-throughput and subjective. AI integrates with high-content imaging and electrophysiology to provide quantitative, high-throughput QC.
Diagram 2: AI-driven phenotypic QC workflow.
Detailed Protocol: AI-Driven Maturity and Toxicity Assessment
- Data Acquisition:
- Imaging: Fix and immunostain hiPSC-CMs for α-actinin to visualize sarcomeres. Acquire high-resolution images using a confocal microscope across multiple wells and batches.
- Electrophysiology: Record extracellular field potentials from hiPSC-CMs plated on multi-electrode arrays (MEAs) before and after exposure with a compound of interest (e.g., a drug suspected of causing arrhythmia) [3].
- AI/ML Processing:
- Sarcomere Analysis: Train a convolutional neural network (CNN) on the stained images using human-annotated datasets for sarcomere organization. The model outputs a quantitative "maturity score" based on Z-disc alignment and sarcomere length regularity.
- Toxicity Prediction: Extract features from MEA recordings, such as field potential duration (FPD), which correlates with the QT interval in vivo. Train a classifier model on historical data from drugs known to cause Torsades de Pointes (TdP) versus safe drugs. The model can then predict the pro-arrhythmic risk of novel compounds [87].
- QC Decision: Establish thresholds for the AI-generated maturity and toxicity scores. Batches of hiPSC-CMs failing to meet the minimum maturity score are excluded from drug screening studies. Compounds flagged as high-risk by the toxicity predictor undergo further, more detailed investigation before proceeding in the development pipeline.
Research Reagent Solutions for Integrated Workflows
The successful implementation of these integrated workflows relies on a suite of specialized reagents and tools.
Table 2: Essential Research Reagents and Tools for CRISPR-AI-iPSC QC
| Reagent / Tool Category | Specific Example | Function in QC Workflow |
|---|---|---|
| CRISPR Editing Tools | Alt-R HDR Enhancer Protein (IDT) | Recombinant protein that improves homology-directed repair (HDR) efficiency during CRISPR editing, increasing the yield of correctly modified isogenic clones [85]. |
| Stem Cell Culture | FUJIFILM Cellular Dynamics iPSCs | Commercially available, well-characterized iPSC lines providing a consistent and reproducible starting material for generating disease models [90]. |
| Delivery Systems | MaxCyte Flow Electroporation | Enables high-efficiency, scalable delivery of CRISPR components (RNPs) into hard-to-transfect iPSCs with maintained cell viability [85]. |
| Differentiation Kits | Commercially available Cardiomyocyte Differentiation Kits | Provide standardized protocols and reagents for generating hiPSC-CMs with consistent purity and yield, reducing batch-to-batch variability. |
| AI/Software Platforms | Yokogawa High-Content Imaging Systems | Automated imaging systems that generate high-dimensional data on cell morphology, ideal for training AI models for phenotypic QC [90]. |
Current Challenges and Future Directions
Despite significant progress, several technical challenges remain for the full integration of CRISPR and AI in iPSC QC. iPSC-CM Immaturity continues to be a major hurdle, as fetal-like cells may not fully recapitulate adult-onset cardiovascular diseases [3]. Future research is focused on advanced maturation techniques, including 3D engineered tissues and prolonged culture, whose outcomes can be quantified by the AI-driven QC metrics described above. CRISPR Off-Target Effects remain a concern, necessitating rigorous validation with high-fidelity Cas variants and extensive NGS in clinical applications [84] [89]. Model Interpretability and Regulatory Alignment for AI tools are also critical. The "black box" nature of some complex AI models requires the development of explainable AI (XAI) to build trust and meet evolving regulatory standards from the FDA and EMA [86] [88].
Looking forward, the field is moving toward more integrated and automated systems. The combination of AI-driven microfluidic systems for cell culture and differentiation, coupled with real-time phenotypic monitoring, will create a closed-loop QC system that continuously optimizes conditions and flags anomalies. Furthermore, the application of these integrated platforms is expanding beyond inherited monogenic diseases to model complex polygenic cardiovascular conditions like hypertension and atrial fibrillation, enabling a new era of personalized drug discovery and safety testing.
Validating hiPSC-CMs: Comparative Analysis with Traditional Models
The pursuit of novel cardiovascular therapeutics is hampered by a persistent and critical translational gap between preclinical animal studies and human clinical outcomes. This whitepaper delineates the fundamental species-specific limitations inherent in traditional animal models of cardiac physiology. Furthermore, it examines how human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and advanced engineered cardiac tissues are emerging as transformative, human-relevant platforms that overcome these limitations. By providing a direct benchmark against human biology, these technologies enhance the accuracy of disease modeling and drug response prediction, thereby de-risking the drug development pipeline.
Cardiovascular disease remains the leading cause of death globally, responsible for approximately 19.8 million deaths annually [3] [27]. Despite this immense burden, the number of new cardiovascular drugs approved each year is steadily decreasing, creating a significant gap between clinical need and therapeutic innovation [3]. A fundamental reason for this low success rate is the lack of preclinical models that can accurately evaluate therapeutic efficacy and safety in humans [3]. For decades, drug development has relied heavily on animal models. However, due to profound differences in cardiac biology between species, these models have a limited ability to predict drug efficacy and cardiotoxicity in humans [3] [91]. The emergence of hiPSC technology provides a paradigm shift. hiPSCs can be differentiated into cardiomyocytes, offering a patient-specific, human-based platform that accurately recapitulates key aspects of human heart biology and disease [38] [91]. This document benchmarks animal models against hiPSC-based systems, highlighting the technical and physiological limitations of the former and outlining how the latter are advancing cardiovascular research and drug discovery.
Fundamental Limitations of Animal Models in Cardiac Physiology
Animal models fail to fully replicate human cardiac pathophysiology due to inherent species-specific differences in genetics, electrophysiology, structure, and metabolism. The following table summarizes these critical disparities.
Table 1: Key Species-Specific Differences in Cardiac Physiology Between Common Animal Models and Humans
| Physiological Parameter | Mouse (Most Common Model) | Human | Implication for Drug Discovery |
|---|---|---|---|
| Heart Rate | ~600 beats per minute [3] | ~60-100 beats per minute [3] | Altered drug kinetics and response; poor prediction of rate-dependent effects. |
| Major Repolarizing K+ Currents | Ito, IK,slow1, IK,slow2, ISS [3] | IKs, IKr [3] | High risk of mispredicting drug-induced arrhythmia (e.g., Torsades de Pointes). |
| Primary Myosin Heavy Chain (MHC) Isoform in Ventricles | α-MHC [3] | β-MHC [3] | Divergent contractile performance and metabolic efficiency. |
| Metabolic Profile | ~90% from mitochondria; well-developed cristae [3] | ~70% from glycolysis; sparse cristae in hiPSC-CMs, improving with maturation [3] | Altered susceptibility to metabolic stress and toxicity. |
| Action Potential Morphology | Very short duration, triangular shape [92] | Long duration with a characteristic plateau [92] | Fundamental difference in electrical activation affects pro-arrhythmia assessment. |
These differences are not merely academic; they have direct, consequential impacts on drug development. For instance, the dependence on different potassium currents for cardiac repolarization is a primary reason why drugs that appear safe in rodent models can provoke lethal arrhythmias in humans [3]. Furthermore, primary human cardiomyocytes obtained from biopsies are not a viable alternative, as they rapidly dedifferentiate and lose their characteristic properties in culture [3]. These limitations create an urgent need for more human-relevant cardiac models.
hiPSC-CMs as a Human-Relevant Benchmarking Platform
hiPSC-CMs represent a revolutionary tool for cardiac research. Somatic cells (e.g., from skin or blood) are reprogrammed into pluripotent stem cells, which are then differentiated into functional cardiomyocytes [38]. This process captures the patient's unique genetic makeup, enabling the creation of "disease-in-a-dish" models.
Advancements in hiPSC-CM Generation and Maturation
A significant challenge has been the immature, fetal-like phenotype of early hiPSC-CMs. Current research focuses on enhancing maturation to better mimic adult human cardiomyocytes (AdCMs).
Table 2: Key Differences Between Immature hiPSC-CMs and Adult Human Cardiomyocytes (AdCMs)
| Characteristic | hiPSC-CMs (Immature) | Adult Human CMs (Mature) |
|---|---|---|
| Cell Morphology | Small, rounded (3000-6000 μm³) [3] | Cylindrical, large (~40,000 μm³) [3] |
| Sarcomere Organization | Poorly organized, random orientation [3] | Highly organized, parallel myofibrils [3] |
| T-Tubules | Rarely observed [3] | Highly developed regular network [3] |
| Calcium Handling | Delayed CICR due to LTCC-RYR2 uncoupling [3] | Rapid, synchronous CICR [3] |
| Metabolic Energy Source | Primarily glycolysis [20] | Primarily oxidative phosphorylation [20] |
| Sarcomeric Protein Isoforms | High MYH6, MLC2a, ssTnI [3] | High MYH7, MLC2v, cTnI (TNNI3) [3] [93] |
Recent protocols have made significant strides in addressing this immaturity. For example, a 2024 study described an optimized stirred suspension bioreactor system for cardiac differentiation that produces hiPSC-CMs (bCMs) with higher purity (~94%), better yield (~1.2 million cells/mL), and more mature functional properties compared to standard monolayer-differentiated CMs (mCMs) [14]. These bCMs also show less batch-to-batch variation, enhancing experimental reproducibility [14]. Other maturation strategies include:
- Directed Maturation of Cardiac Organoids (DM-hCOs): Transient activation of AMPK and ERR signaling drives transcriptomic, proteomic, and functional maturation, increasing the expression of adult sarcomeric proteins like cTnI [93].
- Metabolic Switching: Forcing cells to utilize fatty acids (e.g., palmitate) as an energy source promotes a shift from glycolytic to oxidative metabolism, a hallmark of adult CMs [93].
- Engineering Complexity: The use of 3D engineered heart tissues (EHTs), organ-on-chip systems, and cardiac organoids that incorporate multiple cell types (fibroblasts, endothelial cells) and provide mechanical and electrical stimulation better mimics the native heart microenvironment, driving maturation [20] [91].
Diagram 1: hiPSC-CM Generation and Maturation Workflow. The process involves reprogramming somatic cells, differentiating them into cardiomyocytes, and applying advanced maturation strategies to achieve an adult-like phenotype.
Experimental Applications and Protocols
Disease Modeling: Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
CPVT is a lethal inherited arrhythmia syndrome that is difficult to model in animals due to differences in calcium handling. A seminal study by Maizels et al. modeled CPVT type 2 using hiPSC-CMs derived from a patient with a D307H mutation in the calsequestrin 2 (CASQ2) gene [94].
Experimental Workflow:
- Cell Sourcing: Generate iPSCs from a CPVT2 patient's somatic cells.
- Differentiation: Differentiate iPSCs into cardiomyocytes using standard protocols.
- Phenotyping: Characterize the disease phenotype by simultaneously recording Ca2+ transients and optical action potentials under adrenergic stimulation (e.g., with isoproterenol).
- Observation: CPVT2 hiPSC-CMs exhibited diastolic Ca2+ leakage, delayed afterdepolarizations (DADs), early afterdepolarizations (EADs), and triggered arrhythmias—recapitulating the cellular hallmarks of the disease [94].
- Drug Screening: Test multiple pharmacological compounds (e.g., propranolol, carvedilol, flecainide) at clinically relevant concentrations. The study found that carvedilol stabilized the ryanodine receptor (RyR2), while flecainide suppressed triggered activity [94].
- Clinical Correlation: The drug responses in hiPSC-CMs were congruent with the patient's own clinical response, demonstrating the potential for personalized medicine [94].
Protocol: Stirred Suspension Bioreactor Differentiation
This protocol [14] is designed for efficient, large-scale, and reproducible production of hiPSC-CMs.
Detailed Methodology:
- Input Cell Quality Control: Begin with a quality-controlled master cell bank of hiPSCs. Confirm pluripotency (e.g., >70% SSEA4 expression via FACS) and normal karyotype [14].
- Embryoid Body (EB) Formation: Culture hiPSCs in a stirred bioreactor system that continuously monitors and adjusts temperature, O2, CO2, and pH. Allow cells to spontaneously aggregate into EBs [14].
- Initiation of Differentiation: Precisely when EB diameter reaches 100 µm (typically at 24 hours), initiate mesoderm differentiation by adding the Wnt activator CHIR99021 (7 µM) for 24 hours [14].
- Wnt Inhibition: After a 24-hour gap, add the Wnt inhibitor IWR-1 (5 µM) for 48 hours to promote cardiac specification [14].
- Maturation and Harvest: Maintain cells in suspension culture with metabolic selection media. Contraction is typically observed by differentiation day 5. Cells can be harvested around day 15, achieving high purity (~94% TNNT2+ cells) [14].
- Cryopreservation: Implement controlled freeze and thaw protocols, achieving post-thaw viability >90% [14].
Diagram 2: Signaling Pathway for Directed Maturation. Transient activation of AMPK and ERR signaling drives key aspects of cardiomyocyte maturation, leading to improved function.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for hiPSC-CM Differentiation and Maturation
| Reagent / Tool | Function / Application | Example / Key Detail |
|---|---|---|
| Small Molecule Agonists/Antagonists | Directing differentiation and maturation pathways. | CHIR99021 (Wnt activator), IWR-1 (Wnt inhibitor), MK8722 (AMPK activator), DY131 (ERRβ/γ agonist) [14] [93]. |
| Bioreactor Systems | Scalable, reproducible 3D suspension culture. | Enables controlled monitoring of O2, CO2, pH, and temperature; improves yield and consistency vs. monolayer [14]. |
| Metabolic Maturation Media | Promoting shift to oxidative metabolism. | Supplementation with fatty acids (e.g., palmitate, oleate) [93]. |
| CRISPR-Cas9 System | Genome editing for creating isogenic controls or introducing mutations. | Essential for confirming causality of mutations by creating genetically matched control lines [95] [91]. |
| Heart-Dyno / EHT Platforms | 3D tissue engineering with mechanical load. | Provides auxotonic load, improving sarcomere alignment and contractile function; used for disease modeling and drug testing [20] [93]. |
The limitations of animal models in cardiac physiology—spanning electrophysiology, contractility, and metabolism—are significant and scientifically well-documented. These differences contribute directly to the high attrition rate of cardiovascular drugs. hiPSC-based technologies are no longer just a promising alternative; they are rapidly establishing themselves as an essential, human-relevant benchmark for cardiac disease modeling and drug discovery. The ongoing development of sophisticated protocols for differentiation, maturation, and the creation of complex 3D engineered tissues is steadily closing the translational gap. The integration of these human-based models into the preclinical workflow, alongside traditional models, provides a more comprehensive and predictive platform for understanding human cardiac pathophysiology and accelerating the development of safer, more effective therapies.
{ title }
Comparison with Primary Human Cardiomyocytes: Availability and Functional Fidelity
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have emerged as a transformative tool for cardiovascular disease modeling and drug discovery, offering a solution to the critical shortage of human primary cardiomyocytes (AdCMs). However, hiPSC-CMs exhibit a functionally immature phenotype that differs significantly from adult human cardiomyocytes. This technical guide provides an in-depth comparison of the availability and functional fidelity of these two cell sources, detailing the molecular basis for hiPSC-CM immaturity, presenting protocols to enhance their maturation, and offering a toolkit for their application in cardiovascular research. Understanding these parameters is essential for leveraging hiPSC-CMs effectively in disease modeling and preclinical drug development.
Cardiovascular disease remains the leading cause of death globally, yet the development of new cardiovascular drugs has been hampered by the lack of predictive human preclinical models [3]. Animal models suffer from significant species-specific differences in cardiac biology, limiting their ability to predict human drug responses and toxicity [3] [96]. Primary adult human cardiomyocytes would be the ideal model but are largely inaccessible for routine research due to their limited availability, inability to proliferate in culture, and rapid dedifferentiation after isolation [3] [97]. Human induced pluripotent stem cell-derived cardiomyocytes represent a breakthrough technology, providing an unlimited, patient-specific cell source for disease modeling, drug screening, and regenerative medicine applications [96] [98]. Despite their promise, the functional immaturity of hiPSC-CMs presents a significant challenge that researchers must address to fully realize their potential.
Availability and Sourcing
Primary Human Cardiomyocytes (AdCMs)
- Limited Availability: Sourcing is restricted to donor hearts or tissue specimens from transplant procedures or biopsies, making them rare and difficult to obtain for most research laboratories [97].
- No Expansion Potential: Isolated AdCMs are terminally differentiated and cannot be expanded in culture, severely limiting the cell numbers available for experimentation [99].
- Rapid Dedifferentiation: When placed in culture, AdCMs quickly lose their mature structural and functional characteristics, making long-term studies challenging [3].
hiPSC-Derived Cardiomyocytes (hiPSC-CMs)
- Virtually Unlimited Supply: hiPSCs can be expanded indefinitely in culture, providing a continuous starting material for cardiomyocyte differentiation [96] [20].
- Multiple Sourcing Options: hiPSCs can be reprogrammed from readily accessible somatic tissues including peripheral blood, skin fibroblasts, or even urine epithelial cells [20].
- Patient-Specific Models: The ability to generate hiPSCs from individual patients enables creation of personalized disease models that retain the donor's complete genetic background [96] [98].
Table 1: Comparison of Cardiomyocyte Source Availability
| Parameter | Primary Adult Cardiomyocytes (AdCMs) | hiPSC-Derived Cardiomyocytes (hiPSC-CMs) |
|---|---|---|
| Source Tissue | Donor hearts (transplant/biopsy) | Blood, skin, urine; multiple options |
| Scalability | Severely limited; no expansion | Virtually unlimited; high expansion potential |
| Genetic Diversity | Limited by donor availability | Broad; patient-specific lines possible |
| Regulatory Concerns | Ethical considerations for tissue use | Minimal ethical concerns; established guidelines |
Functional Fidelity: Structural and Molecular Characteristics
Cell Morphology and Ultrastructure
Substantial structural differences exist between hiPSC-CMs and adult cardiomyocytes, representing a fundamental aspect of their functional disparity:
- Cell Shape and Size: AdCMs exhibit a characteristic rod-shaped morphology with dimensions of approximately 140 µm in length and 20 µm in width (volume ~40,000 µm³). In contrast, hiPSC-CMs are smaller and more rounded, with volumes of only 3,000-6,000 µm³ [3].
- Sarcomeric Organization: AdCMs contain highly organized, parallel myofibrils with sarcomere lengths of 1.9-2.2 µm. hiPSC-CMs display disorganized, randomly oriented sarcomeres with shorter lengths (1.7-2.0 µm) [3].
- Sarcomere Protein Isoforms: The sarcomeres of AdCMs and hiPSC-CMs express different protein isoforms, reflecting their distinct maturation states:
- Myosin Heavy Chain (MHC): AdCMs express predominantly βMHC, while hiPSC-CMs express the fetal αMHC isoform [3].
- Myosin Light Chain (MLC): AdCMs express MLC2v, while hiPSC-CMs predominantly express the fetal MLC2a isoform [3].
- Troponin I (TnI): AdCMs express cardiac TnI, while hiPSC-CMs express slow-twitch skeletal TnI (ssTnI) [3].
- T-Tubule Development: AdCMs possess a well-developed T-tubule network essential for coordinated calcium-induced calcium release. hiPSC-CMs largely lack T-tubules, leading to uncoupling of L-type calcium channels and ryanodine receptors (RYR2) and consequently impaired calcium handling [3].
Electrophysiological Properties and Calcium Handling
Functional differences in electrophysiology and calcium handling significantly impact the utility of hiPSC-CMs for drug safety testing and disease modeling:
- Spontaneous Contraction: hiPSC-CMs exhibit spontaneous beating activity similar to neonatal cardiomyocytes, while AdCMs require external electrical stimulation to contract, reflecting differences in their underlying pacemaker currents and ion channel expression [97].
- Ion Channel Expression: Both the absolute and relative expression levels of critical ion channels differ between AdCMs and hiPSC-CMs, with hiPSC-CMs often showing under- or over-expression of key channels compared to the physiological levels in AdCMs [97].
- Excitation-Contraction Coupling: The calcium handling machinery in hiPSC-CMs is functionally immature, exhibiting delayed calcium transients and aberrant inotropic responses to pharmacological agents compared to AdCMs [97].
- Action Potential Characteristics: hiPSC-CMs typically show shorter action potential duration and altered morphology compared to AdCMs, reflecting differences in the complement and regulation of repolarizing potassium currents [3] [76].
Table 2: Functional Comparison of Cardiomyocyte Types
| Functional Parameter | Adult Human Cardiomyocytes | hiPSC-Derived Cardiomyocytes |
|---|---|---|
| Cell Morphology | Rod-shaped, organized | Irregular, rounded |
| Sarcomeric Structure | Highly organized, aligned | Disorganized, random orientation |
| T-Tubule Network | Abundant, well-organized | Deficient or absent |
| Contraction Pattern | Requires pacing | Spontaneous |
| Ion Channel Expression | Physiological levels | Often under- or over-expressed |
| Calcium Handling | Mature, rapid transients | Immature, delayed transients |
| Force-Frequency Relationship | Positive | Negative (immature) |
| Metabolic Profile | Predominantly fatty acid β-oxidation | Predominantly glycolysis |
Experimental Approaches to Enhance hiPSC-CM Maturity
Advanced Differentiation Protocols
Traditional hiPSC-CM differentiation protocols utilize broad-spectrum pharmacological inhibitors that typically yield immature cardiomyocytes. Recent advances have focused on more specific molecular approaches:
- Sfrp2-Based Differentiation: Replacement of broad-spectrum Wnt inhibitors with Secreted Frizzled-Related Protein 2 (Sfrp2), a specific Wnt3a inhibitor, produces hiPSC-CMs with enhanced structural and functional maturity [76]. This method results in:
- Longer sarcomeres with improved organization
- Reduced cellular circularity (more adult-like morphology)
- Longer action potential duration
- Formation of polarized gap junctions between cardiomyocytes
- Lower spontaneous beating rates, more similar to adult cardiomyocytes [76]
Diagram 1: Sfrp2 differentiation protocol workflow (Title: Enhanced Cardiac Differentiation via Sfrp2)
- Lineage-Specific Reprogramming: hiPSCs generated from cardiac-derived fibroblasts (as opposed to dermal fibroblasts or blood cells) retain epigenetic memory that promotes more efficient cardiac differentiation and yields hiPSC-CMs with more adult-like calcium handling and electrophysiological profiles [99].
Tissue Engineering and Maturation Strategies
Beyond differentiation protocol improvements, various tissue engineering approaches can enhance hiPSC-CM maturation:
- Three-Dimensional Engineered Tissues: Culturing hiPSC-CMs in 3D hydrogel constructs provides biomechanical cues and enhanced cell-cell interactions that promote structural and functional maturation, including improved sarcomeric organization and more adult-like electrophysiology [26] [20].
- Mechanical Loading: Application of controlled mechanical stress to hiPSC-CMs or engineered tissues mimics the physiological loading conditions of the native heart and promotes hypertrophic growth and maturation through integrin-mediated mechanotransduction pathways [26].
- Electrical Pacing: Chronic electrical stimulation at physiologically relevant frequencies (1-2 Hz) can enhance hiPSC-CM maturation by promoting electromechanical coupling, improving calcium handling, and driving the development of a more adult-like electrophysiological phenotype [20].
- Metabolic Maturation: Shifting the culture medium from glucose-rich to fatty acid-containing media forces hiPSC-CMs to utilize fatty acid β-oxidation as their primary energy source, mimicking the metabolic profile of adult cardiomyocytes and promoting overall maturation [76].
Diagram 2: hiPSC-CM maturation strategies (Title: hiPSC-CM Maturation Approaches)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for hiPSC-CM Differentiation and Maturation
| Reagent Category | Specific Examples | Function in Cardiac Differentiation/Maturation |
|---|---|---|
| Small Molecule Inhibitors | CHIR99021 (GSK3 inhibitor), IWR-1, Wnt-C59, XAV939 (Wnt inhibitors) | Direct differentiation toward cardiac lineage by modulating Wnt/β-catenin signaling [30] [76] |
| Growth Factors & Cytokines | BMP4, Activin A, FGF2, VEGF | Recapitulate developmental signaling pathways to promote cardiac mesoderm formation and patterning [30] |
| Maturation Factors | Sfrp2, Triiodothyronine (T3), Dexamethasone, Fatty acids | Promote structural and functional maturation through specific signaling pathways and metabolic manipulation [76] |
| Culture Matrices | Matrigel, Geltrex, Laminin-521, Synthemax II-SC | Provide substrate for cell attachment and survival, influencing differentiation efficiency and maturation [30] |
| Culture Media | RPMI/B27, Defined media (E8, B8), Fatty acid-supplemented media | Support cell survival while providing specific induction or maturation cues [30] [76] |
hiPSC-derived cardiomyocytes represent a powerful alternative to primary adult cardiomyocytes for cardiovascular research, primarily due to their unlimited availability and patient-specific nature. However, their functional immaturity compared to adult cardiomyocytes remains a significant limitation that researchers must address through advanced differentiation protocols and maturation strategies. The continued development of methods to enhance hiPSC-CM maturity, particularly through specific Wnt pathway modulation via Sfrp2 and tissue engineering approaches, is bringing these cells closer to faithfully recapitulating adult human cardiac physiology. As these technologies evolve, hiPSC-CMs will play an increasingly central role in disease modeling, drug discovery, and the development of personalized medicine approaches for cardiovascular disorders.
The translation of induced pluripotent stem cell (iPSC) technology from basic research to clinical application represents a paradigm shift in cardiovascular medicine. This whitepaper delineates the critical pathway linking iPSC-derived cardiovascular disease models with clinical trial outcomes and patient data, underscoring their growing role in de-risaging drug development and validating therapeutic efficacy. We provide a systematic analysis of current clinical trial landscapes, detailed protocols for generating clinically relevant iPSC-cardiomyocytes (iPSC-CMs), and advanced computational frameworks for correlating in vitro findings with patient-specific responses. Within the broader thesis of iPSC applications in cardiovascular research, this document establishes a foundational roadmap for researchers and drug development professionals to enhance the predictive validity and clinical translatability of iPSC-based platforms.
Cardiovascular diseases remain a leading cause of global morbidity and mortality, yet the drug development pipeline has been hampered by high attrition rates, often due to the poor predictive power of traditional animal models and immortalized cell lines for human cardiotoxicity and efficacy [100]. The emergence of human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) has created unprecedented opportunities for patient-specific disease modeling, drug screening, and regenerative therapy development. The core translational relevance of these platforms lies in their ability to bridge the critical gap between experimental findings and clinical outcomes by preserving the human genetic context and enabling direct correlation with patient data [101] [100]. This document examines the evidence supporting this correlation, details the methodologies that ensure clinical fidelity, and outlines the integrative approaches that are refining the predictive power of iPSC-based cardiovascular research.
Clinical Trial Landscape: From iPSC Models to Patient Outcomes
A systematic assessment of the clinical trial landscape is essential to validate the translational promise of iPSC technology. A recent scoping review of published clinical studies and registered trials provides a quantitative snapshot of this rapidly evolving field [18].
Table 1: iPSC-Based Clinical Trials and Studies (as of January 2025)
| Category | Number | Total Patients Treated (Published Studies) | Key Disease Areas |
|---|---|---|---|
| Published Clinical Studies | 10 | 115 | Cardiac conditions, ocular disorders, cancer, graft-versus-host disease [18] |
| Ongoing Registered Trials | 22 | Not Applicable | Cardiac conditions, ocular disorders, cancer, graft-versus-host disease, platelet transfusions [18] |
| Notable Characteristics | Studies are predominantly small and uncontrolled; only two published studies enrolled more than four patients. |
The data reveals that iPSC-based interventions are being actively explored for a wide range of conditions. In cardiology, early trials have pioneered the use of iPSC-derived cell products. For instance, a landmark trial in Japan under Dr. Yoshiki Sawa involved transplanting thin wafer-like sheets of iPSC-derived cardiomyocytes onto damaged myocardium in patients with heart failure [102]. Preclinical data leading to this trial demonstrated safety and functional improvement in porcine models, primarily through paracrine release of cytokines and growth factors [102]. However, this trial also highlights existing challenges, including the need for immunosuppression and the difficulty of assessing efficacy without randomized control groups [102]. The small scale and uncontrolled design of most current studies, as identified in the scoping review, preclude definitive conclusions about efficacy, underscoring the need for larger, controlled trials and standardized outcome reporting [18].
Experimental Protocols: Generating Clinically Relevant iPSC-Cardiomyocytes
The correlation between in vitro models and clinical data hinges on the reproducibility, scalability, and maturity of the iPSC-CMs. Traditional monolayer differentiation faces challenges in batch-to-batch consistency and scale. The following optimized protocol for stirred suspension systems addresses these limitations, enhancing translational relevance by producing high-quality, functional cardiomyocytes suitable for disease modeling and drug screening [14].
Optimized Bioreactor Protocol for iPSC-Cardiomyocyte Differentiation
Objective: To efficiently generate a high yield of pure, ventricular-like, and functionally mature human iPSC-derived cardiomyocytes (hiPSC-CMs) in a stirred suspension bioreactor system.
Key Advantages Over Monolayer Differentiation:
- Enhanced Reproducibility: Reduced well-to-well and batch-to-batch variation.
- Superior Scalability: Linear scaling from small spinner flasks to large bioreactors.
- Improved Functional Properties: bCMs exhibit more mature functional properties compared to monolayer-differentiated CMs (mCMs) [14].
- Robust Cryopreservation: High viability (>90%) post-thaw, facilitating experimental logistics [14].
Materials and Reagents:
- Quality-Controlled hiPSC Master Cell Bank: Karyotypically normal and mycoplasma-free.
- Stirred Bioreactor System: Capable of monitoring and adjusting temperature, O~2~, CO~2~, and pH.
- Small Molecule Inhibitors and Activators: CHIR99021 (Wnt activator), IWR-1 (Wnt inhibitor).
- Differentiation and Culture Media: As defined in the protocol.
Methodology:
- Quality Control of Input hiPSCs: Ensure pluripotency marker SSEA4 expression is >70% by FACS analysis. Lower levels predispose differentiation to failure [14].
- Formation of Embryoid Bodies (EBs): Transfer hiPSCs to suspension culture to allow aggregation and EB formation. Monitor EB diameter closely.
- Initiation of Mesoderm Differentiation (Day 0): Add 7 µM CHIR99021 to activate Wnt signaling. Critical Step: Add CHIR only when the majority of EBs reach a diameter of 100 µm. EBs smaller than 100 µm may disintegrate, while those larger than 300 µm differentiate less efficiently due to diffusion limits [14].
- Wnt Inhibition (Day 2): 24 hours after CHIR addition, replace the medium to remove CHIR. 24 hours after that (48 hours post-CHIR addition), add 5 µM IWR-1 to inhibit Wnt signaling for 48 hours.
- Maturation and Maintenance: Continue culture in cardiac maintenance medium, with medium changes every 2-3 days. Spontaneous contractions are typically observed by differentiation day 5 [14].
- Harvesting and Cryopreservation: Harvest bCMs from day 15 onwards using gentle enzymatic dissociation. Use controlled freeze and thaw protocols to maintain high viability and functional properties.
Table 2: Benchmark Outcomes for Bioreactor-Differentiated iPSC-CMs
| Parameter | Benchmark Outcome | Comparison to Monolayer (mCMs) |
|---|---|---|
| Average Yield | ~1.21 million cells per mL [14] | Lower [14] |
| Purity (%TNNT2+) | ~94% [14] | More variable [14] |
| Viability Post-Thaw | >90% [14] | Often lower; functional properties more affected [14] |
| Ventricular Identity | 83.4% MLC2v+ by flow cytometry; high expression of MYH7, MYL2, MYL3 [14] | Less pronounced |
| Onset of Contraction | Differentiation Day 5 [14] | Differentiation Day 7 [14] |
Workflow Diagram: Bioreactor Differentiation of iPSC-CMs
The following diagram illustrates the optimized protocol for generating iPSC-derived cardiomyocytes in a stirred suspension system.
The Scientist's Toolkit: Essential Reagents and Platforms
Successful implementation of clinically relevant iPSC-based research requires a suite of specialized reagents and platforms. The following table details key solutions for cardiovascular disease modeling.
Table 3: Research Reagent Solutions for iPSC-Based Cardiovascular Modeling
| Item | Function/Application | Technical Notes |
|---|---|---|
| Small Molecule Wnt Activator (CHIR99021) | Initiates mesoderm differentiation by activating Wnt/β-catenin signaling. | Concentration and timing are critical; typically used at 7 µM for 24h in suspension systems [14]. |
| Small Molecule Wnt Inhibitor (IWR-1) | Promotes cardiac specification by inhibiting Wnt signaling after mesoderm formation. | Used at 5 µM for 48h in optimized protocols; more cost-effective than growth factors [14]. |
| Stirred Suspension Bioreactor | Provides a controlled, scalable environment for differentiation; improves nutrient distribution and reduces batch variation. | Enables monitoring of O~2~, CO~2~, pH, and temperature [14]. |
| Multi-Electrode Array (MEA) | Functional screening of electrophysiological properties in iPSC-CM networks. | Detects pro-arrhythmic phenotypes and drug effects on field potential [103]. |
| Cardiac Organoid & 3D Culture Platforms | Generate complex, self-organizing 3D tissue models for studying structural diseases and maturation. | Used for modeling ventricular wall formation, congenital diseases, and drug cardiotoxicity [104] [105]. |
| Reprogramming & Differentiation Platforms | Core technology for generating patient-specific iPSCs and directing their differentiation into cardiomyocytes. | Essential for creating large-scale, consistent cell sources for disease modeling and drug screening [105]. |
Advanced Correlation: Computational Integration of iPSC and Clinical Data
Bridging the gap between in vitro iPSC models and human clinical outcomes increasingly relies on sophisticated computational approaches. Two key methodologies are enhancing this correlation:
Simulation-Based Inference (SBI) for Mechanistic Insight
Machine learning techniques, particularly Simulation-Based Inference (SBI), are being applied to infer underlying disease mechanisms from the electrical activity of patient-derived neuronal and cardiac networks recorded on Multi-Electrode Arrays (MEAs) [103]. SBI uses a biophysically detailed computational model to simulate network activity. By training a deep neural network on millions of simulations, it can analyze experimental MEA data from patient-derived cells and calculate the posterior probability of all possible model parameters that could explain the observation [103]. This allows researchers to pinpoint specific altered biological properties (e.g., ion channel conductances, synaptic strengths) in diseased networks, directly linking patient-specific cellular phenotypes to potential molecular mechanisms.
Large Language Models (LLMs) for Data Integration and Hypothesis Generation
Large Language Models (LLMs) like GPT-4 and BioGPT are emerging as powerful tools to accelerate iPSC-CM research and its clinical translation. Their applications include:
- Literature Mining: Identifying novel gene-phenotype relationships and potential therapeutic targets from the vast biomedical literature far more efficiently than traditional methods [106].
- Protocol Optimization: Assisting in the design and refinement of complex CRISPR-based gene editing and differentiation protocols [106].
- Multi-Omics Integration: Merging patient-level genomic, transcriptomic, and clinical data with high-content functional outputs from iPSC-CMs to uncover personalized disease signatures and predict patient-specific drug responses [106].
The following diagram illustrates how these computational tools integrate with experimental and clinical data streams to enhance translational prediction.
The translational relevance of iPSC technology in cardiovascular disease modeling is rapidly maturing from a promising concept to a tangible tool with direct correlations to clinical trial outcomes and patient data. The convergence of robust, scalable differentiation protocols like stirred suspension bioreactors, advanced 3D organoid models, and powerful computational intelligence from SBI and LLMs creates a synergistic framework. This framework enables a more predictive, patient-centric approach to drug discovery and safety assessment. While challenges in standardization, functional maturation, and validation in large-scale controlled trials remain, the strategic integration of these elements is decisively closing the loop between the laboratory bench and the patient bedside, paving the way for a new era in cardiovascular regenerative medicine and therapeutic development.
The adoption of induced pluripotent stem cells (iPSCs) represents a paradigm shift in biomedical research and therapeutic development. Since their discovery by Takahashi and Yamanaka in 2006, iPSCs have emerged as a powerful platform for disease modeling, drug discovery, and regenerative medicine [77]. The pharmaceutical and biotechnology sectors are increasingly leveraging iPSC-derived cardiomyocytes (iPSC-CMs) to model cardiovascular diseases with unprecedented human relevance, overcoming the limitations of traditional animal models that often fail to accurately predict human physiology and drug responses [3] [60]. This technical guide examines the current state of iPSC adoption, with a specific focus on their application in cardiovascular disease modeling, detailing market drivers, experimental methodologies, and the essential toolkit for researchers.
The iPSC market is experiencing robust growth, fueled by rising R&D investment and expanding therapeutic applications. The global induced pluripotent stem cells market size is projected to increase from USD 2.13 billion in 2025 to approximately USD 5.12 billion by 2034, representing a compound annual growth rate (CAGR) of 10.25% [107]. Another analysis projects the market will reach USD 4.69 billion by 2033, growing at a CAGR of 9.86% from 2024 [108].
Table 1: Global iPSC Market Size Projections
| Market Size 2024/2025 | Projected Market Size | CAGR | Source |
|---|---|---|---|
| USD 2.13 billion (2025) | USD 5.12 billion (2034) | 10.25% | [107] |
| USD 2.01 billion (2024) | USD 4.69 billion (2033) | 9.86% | [108] |
| USD 1.92 billion (2025) | USD 4.34 billion (2034) | 9.5% | [109] |
The drug discovery and development segment dominates the application landscape, accounting for approximately 40.23% of 2024 market revenue [110]. This segment's leadership position is attributed to the pharmaceutical industry's rapid adoption of iPSC-derived cells for high-throughput drug screening and toxicity testing.
Table 2: iPSC Market Share by Application and End User (2024)
| Segment | Leading Category | Market Share | Fastest-Growing Category | Growth Rate |
|---|---|---|---|---|
| Application | Drug Discovery & Development | 40.23% [110] | Regenerative Medicine | 12.43% CAGR [110] |
| End User | Pharmaceutical & Biotechnology Companies | 58.79% [110] | Academic & Research Institutions | 12.85% CAGR [110] |
| Derived Cell Type | Cardiomyocytes | 28.94% [110] | Neurons | 11.13% CAGR [110] |
Geographically, North America dominated the global market with a 36-37.51% share in 2024, supported by substantial NIH funding, an active venture capital ecosystem, and progressive FDA guidance on alternative methods [107] [110]. However, the Asia-Pacific region is projected to be the fastest-growing market, with a CAGR of 12.32% to 2030, buoyed by Japan's fast-track approvals and significant sovereign funding [110].
Key Adoption Drivers in Pharma and Biotech
Regulatory Shifts and Human-Relevant Models
The 2024 FDA guidance formally accepted iPSC platforms for toxicology submissions, providing developers with crucial regulatory clarity and accelerating pharmaceutical adoption [110]. This regulatory shift comes as the limitations of animal models become increasingly apparent – fundamental differences in cardiac biology between species limit their ability to predict human drug responses [3]. For instance, mouse heart rates are approximately eight times faster than humans, and cardiac repolarization depends on different ion currents [3]. iPSC-CMs address these limitations by providing human-specific cardiac cells that more accurately predict drug efficacy and cardiotoxicity in humans.
Economic Imperatives in Drug Development
The economic rationale for iPSC adoption is compelling. The drug development success rate remains low, with only approximately 5% of new molecular entities (NMEs) ultimately approved [3]. Each late-stage failure avoided can save over USD 100 million in sunk costs, creating a powerful incentive for implementing more predictive human iPSC models early in the drug development pipeline [110]. Pharmaceutical companies are increasingly using purpose-built iPSC-derived cardiomyocyte kits, such as atrial cardiomyocyte panels for arrhythmia evaluation, to match specific preclinical endpoints and reduce assay variability [110].
Technological Advancements in iPSC Production
Continuous improvements in reprogramming technologies, differentiation protocols, and automation have strengthened the scalability and reliability of iPSC production. Automated closed-system bioreactors can lower labor needs by 70% and reduce per-batch costs by 50% [110]. High-throughput microfluidic sorters can now remove undifferentiated cells at rates of 3 million cells/minute, enhancing the safety profiles of iPSC-derived products [110]. These advancements have enabled suppliers like FUJIFILM Cellular Dynamics to scale daily output to billions of iPSC-derived cells for drug screening applications [110].
iPSCs in Cardiovascular Disease Modeling: Experimental Framework
Protocol for Directed Cardiac Differentiation
The following workflow outlines the established methodology for generating cardiomyocytes from iPSCs using a monolayer-based, small molecule-directed differentiation approach, which can achieve efficiencies of up to 95% [60]:
Diagram: Cardiac Differentiation from Human iPSCs. This workflow outlines the key stages and signaling pathways involved in generating functional cardiomyocytes from iPSCs, highlighting critical timepoints and factors.
The differentiation process recapitulates mammalian heart development, progressing through mesoderm formation, cardiac mesoderm specification, and finally cardiomyocyte differentiation [60]. Early protocols using embryonic body (EB) spontaneous differentiation yielded only about 5% cardiomyocytes, but contemporary monolayer-based, small molecule-directed differentiation methods can generate >95% pure populations of iPSC-derived cardiomyocytes (iPSC-CMs) in a cost-effective manner [60].
Chamber-Specific Cardiomyocyte Differentiation
A significant advancement in the field has been the development of protocols for generating chamber-specific cardiomyocytes. Different forms of heart disease target specific cardiac regions – for example, long QT syndrome primarily affects the left ventricle, while Brugada syndrome mainly impacts the right ventricle [60]. Researchers have successfully derived human ventricular and atrial cardiomyocytes from different mesoderm populations identified by CD235a and RALDH2 expression in the early stages of differentiation [60]. More recently, double reporter lines (e.g., TBX5Clover2/NKX2-5TagRFP) have enabled isolation of first heart field (FHF) and second heart field (SHF)-derived iPSC-CMs, allowing for more precise modeling of region-specific cardiac diseases [60].
Maturation Strategies to Overcome Functional Limitations
A major challenge in utilizing iPSC-CMs for disease modeling is their immature phenotype, which resembles fetal rather than adult cardiomyocytes [3] [60]. Several maturation strategies have been developed:
Structural maturation: Adult cardiomyocytes have a cylindrical shape (∼40,000 μm³) with well-organized sarcomeres, while iPSC-CMs are smaller (3,000-6,000 μm³) with randomly oriented sarcomeres [3]. Maturation protocols aim to promote sarcomere organization and T-tubule development through prolonged culture and electrical stimulation.
Metabolic maturation: iPSC-CMs primarily rely on glycolysis for energy production, similar to fetal cardiomyocytes, while adult cardiomyocytes depend on mitochondrial oxidative phosphorylation of fatty acids [3]. Maturation approaches include using fatty acid-rich media to promote metabolic switching.
Electrophysiological maturation: The development of T-tubules is crucial for proper calcium handling. Immature iPSC-CMs show delayed calcium-induced calcium release due to spatial uncoupling between L-type Ca2+ channels and RYR2 [3]. Prolonged culturing, electrical pacing, and 3D culture systems promote the formation of these critical structures.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for iPSC-Cardiomyocyte Research
| Category | Specific Examples | Function & Application |
|---|---|---|
| Reprogramming Kits | Sendai virus vectors, episomal plasmids, mRNA kits | Reprogram somatic cells (fibroblasts, blood cells) to pluripotent state with minimal genomic integration risk [111] [110] |
| Cardiac Differentiation Kits | Commercially available differentiation kits (e.g., Gibco, STEMCELL Technologies) | Standardized protocols and media formulations for efficient, reproducible cardiac differentiation [110] |
| Cell Culture Consumables | Matrigel, laminin-521, vitronectin; defined culture media | Provide extracellular matrix support for iPSC expansion and maintenance under xeno-free conditions [111] |
| Characterization Reagents | Flow cytometry antibodies (cTnT, NKX2-5), PCR arrays, immunocytochemistry kits | Quality control assessment of pluripotency markers, cardiac differentiation efficiency, and purity [111] [60] |
| Automated Platforms | Closed-system bioreactors, high-throughput microfluidic sorters | Enable scalable, reproducible GMP-grade production; reduce labor needs by 70% and costs by 50% [111] [110] |
Applications in Cardiovascular Disease Modeling and Drug Discovery
Inherited Arrhythmia Syndromes
iPSC-CMs have proven particularly valuable for modeling inherited arrhythmic syndromes such as Long QT Syndrome (LQTS), Brugada Syndrome, and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) [60]. These conditions often involve ion channel mutations that manifest differently in human cells compared to animal models. For instance, researchers studying LQT2 with a hERG1 mutation (H70R) identified a complex phenotype involving hERG1a and hERG1b ratio imbalance in addition to mutant channel trafficking abnormalities – nuances not apparent in heterologous expression systems [60].
Drug Screening and Cardiotoxicity Testing
The integration of iPSC-CMs into drug development pipelines has transformed early-stage safety pharmacology. The following workflow illustrates a typical drug screening platform using iPSC-derived cardiomyocytes:
Diagram: Drug Screening Platform Using iPSC-Cardiomyocytes. This workflow demonstrates the process from patient cell acquisition through functional screening, highlighting the human-relevant platform for drug discovery.
This approach enables researchers to study patient-specific responses to drug compounds and accurately assess cardiotoxicity risks before advancing to clinical trials. The platform combines robotic plate handlers, optical mapping, and calcium-flux analytics with standardized iPSC panels to expedite hit-to-lead workflows [110].
Challenges and Future Directions
Despite significant progress, several challenges remain in the widespread adoption of iPSC technology for cardiovascular applications. Tumorigenicity concerns related to residual undifferentiated iPSCs necessitate rigorous purification processes [18] [77]. The high production costs and limited scalability of GMP-grade iPSC production continue to hinder broader clinical translation [107] [110]. Furthermore, complex and evolving global regulatory frameworks for cell-based therapeutics create uncertainty and delay commercialization timelines [110].
Future directions focus on enhancing cardiomyocyte maturation through advanced 3D culture systems, organoid development, and vascularization [112]. The integration of artificial intelligence and machine learning for image analysis and predictive modeling is also accelerating, shrinking manual workloads and cycle times [107] [110]. As these technologies mature and standardization improves, iPSC-based cardiovascular disease models are poised to become indispensable tools for drug discovery and personalized medicine approaches.
The integration of human induced pluripotent stem cell (iPSC)-derived cardiomyocytes into drug development pipelines represents a paradigm shift in cardiovascular research. This whitepaper details the current regulatory landscape, underscored by the FDA's push for New Approach Methodologies (NAMs), and the experimental frameworks essential for validating these models. As regulatory bodies move to reduce reliance on animal testing, iPSC-derived systems offer a human-relevant platform for evaluating efficacy and safety from discovery to Investigational New Drug (IND) application. This document provides a technical guide for researchers and drug development professionals to future-proof their cardiovascular disease models by aligning with evolving regulatory expectations and leveraging advanced in vitro methodologies.
The Evolving Regulatory Landscape for iPSC-Based Models
The drug development landscape is undergoing a major transformation driven by regulatory innovation. Regulatory bodies like the FDA are actively promoting a shift away from traditional animal models toward more predictive, human-based systems [90]. This shift is embodied in the adoption of New Approach Methodologies (NAMs), which include advanced in vitro systems, high-content imaging, and computational tools [90].
iPSC-derived cardiomyocytes (iPSC-CMs) are at the forefront of this shift, serving as a core component of biologically relevant NAMs. Their human origin provides unprecedented insights into human cardiac biology and pathology, bridging the translational gap between preclinical research and clinical outcomes. The regulatory pathway is increasingly favoring these models for their ability to improve the predictivity of target identification, drug efficacy, and safety assessments before a candidate reaches clinical trials [90] [36]. This is particularly critical in cardiology, where species-specific differences in cardiac electrophysiology and contractility have led to the failure of compounds that were successful in animal models [3].
Diagram: The Evolving Regulatory and Drug Development Pathway
Status of Clinical Trials and Major Research Initiatives
While large-scale, late-phase clinical trials for iPSC-based cardiac regenerative therapies are still maturing, the research infrastructure supporting drug discovery and safety assessment is advancing rapidly. The current landscape is characterized by foundational research and early-stage initiatives that are building the necessary credibility for regulatory acceptance. Key developments include:
- Preclinical Disease Modeling: Extensive use of patient-specific iPSC-CMs to model monogenic cardiac disorders like Hypertrophic Cardiomyopathy (HCM), which is primarily caused by mutations in sarcomeric genes such as MYH7 and MYBPC3 [40]. These models recapitulate disease phenotypes in vitro, providing platforms for mechanistic studies and drug screening.
- Drug Safety and Toxicity Screening: iPSC-CMs are being integrated into pharmaceutical workflows for cardiotoxicity assessment, particularly for off-target effects of novel therapies like engineered cell therapies [90]. This application addresses a critical need in the drug development pipeline.
- High-Content Phenotyping: Research initiatives are increasingly leveraging high-content imaging and functional assays to characterize iPSC-CM models in depth. This includes detailed measurements of cellular morphology, contractility, electrophysiological properties, calcium handling, and mitochondrial function [40].
- Focused Summit and Collaborations: Events like the 2025 iPSC Summit, featuring leaders from industry and academia, are dedicated to "Integrating New Approach Methodologies with Human iPSCs from Discovery to IND," highlighting the concerted effort to translate research into regulated applications [90].
Table: Key Areas of Active iPSC-CM Research and Application
| Research Focus Area | Key Objective | Example Pathologies/Applications |
|---|---|---|
| Inherited Cardiomyopathy Modeling | Elucidate pathogenesis of genetic disorders; personalized drug testing. | Hypertrophic Cardiomyopathy (HCM), Dilated Cardiomyopathy (DCM) [40]. |
| Drug-Induced Cardiotoxicity | Assess compound safety and off-target effects on human cardiomyocytes. | Preclinical safety screening for chemotherapeutics, novel cell therapies [90] [36]. |
| Protocol Standardization | Improve maturity, reproducibility, and scalability of iPSC-CMs. | Maturation protocols, defined culture media, biophysical stimulation [30] [3]. |
| Precision Medicine & Drug Discovery | Identify patient-specific therapies and discover new mechanism-based drugs. | Patient stratification, high-throughput drug screening [96] [36]. |
Detailed Experimental Protocols for Model Validation
Protocol: Overcoming Contact Inhibition with IGFBP2
A recent breakthrough identified contact inhibition as a primary barrier to cardiomyocyte proliferation and discovered Insulin-like Growth Factor Binding Protein 2 (IGFBP2) as a key mitogen to overcome it [113].
Workflow Overview:
- Cell Model: Use induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs).
- Modeling Contact Inhibition: Culture iPSC-CMs at both sparse (low density) and dense (high density, cell-to-cell contact) conditions.
- Protein Analysis: Analyze secreted proteins from sparse vs. dense cultures. This revealed IGFBP2 is highly secreted by sparse, dividing cells but not by dense, non-dividing cells [113].
- Functional Validation:
- Add recombinant IGFBP2 protein to densely plated, non-dividing iPSC-CMs.
- Assess cell division rates using imaging techniques.
- Validate findings in 3D heart tissue spheroids using tissue clearing and light-sheet microscopy to visualize deep tissue division [113].
Mechanistic Insight: Cell-to-cell contact suppresses the Wnt signaling pathway and causes premature reassembly of sarcomeres, halting division. IGFBP2 reintroduction reactivates the proliferative program [113].
Diagram: Experimental Workflow for IGFBP2 Proliferation Study
Protocol: Enhancing Cardiac Differentiation by Inhibiting Ferroptosis
A major challenge in generating iPSC-CMs is cell death during differentiation. A 2024 study identified ferroptosis as the primary cell death modality in the first 48 hours of differentiation [114].
Workflow Overview:
- Differentiation Initiation: Begin standard cardiac differentiation of human PSCs via temporal modulation of the Wnt pathway (e.g., GSK-3 inhibition).
- Cell Death Analysis: Observe substantial cell detachment and death during the initial stage. Pharmacologically target different cell death pathways (e.g., apoptosis, necrosis, ferroptosis).
- Identification of Ferroptosis: Discover that inhibitors of ferroptosis, but not other death pathways, significantly reduce cell death.
- Protocol Enhancement: Add the ferroptosis inhibitor Ferrostatin-1 (FER-1) to the differentiation medium for the first 48-96 hours.
- Outcome Assessment: Results in increased robustness, cell yield, and efficiency of cardiomyocyte generation [114].
The Scientist's Toolkit: Essential Research Reagents
Success in iPSC-based cardiovascular disease modeling relies on a suite of well-defined reagents and protocols. The following table details key components for culture, differentiation, and experimentation.
Table: Essential Reagents for iPSC Culture, Cardiac Differentiation, and Experimentation
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Basal Culture Media | Essential 8 (E8), B8, TeSR1 | Chemically defined media for robust maintenance of pluripotency in hiPSCs [30]. |
| Extracellular Matrix | Growth-Factor Reduced Matrigel, Geltrex, Synthemax II-SC | Provides a substrate for hiPSC attachment and growth. A 1:800 dilution is often sufficient [30]. |
| Cardiac Differentiation Factors | CHIR99021 (GSK-3 inhibitor), IWP-2/Wnt-C59 (Wnt inhibitor), BMP4, Activin A | Temporal modulation of Wnt and other signaling pathways to direct mesoderm and cardiac lineage specification [30] [114]. |
| Specialized Additives | Ferrostatin-1 (FER-1), Y-27632 (ROCK inhibitor) | FER-1: Inhibits ferroptosis to improve differentiation efficiency [114]. Y-27632: Enhances cell survival after passaging [30]. |
| Key Assay Reagents | Recombinant IGFBP2 | Used to stimulate proliferation in dense cardiomyocyte cultures and 3D spheroids to overcome contact inhibition [113]. |
Future-proofing cardiovascular disease models requires a strategic integration of advanced iPSC technologies within the evolving regulatory framework of New Approach Methodologies. By adopting robust, validated experimental protocols—such as leveraging IGFBP2 to probe proliferation mechanisms or inhibiting ferroptosis to enhance differentiation efficiency—researchers can build more predictive and human-relevant platforms. The ongoing transition from animal models to human iPSC-based systems promises to de-risk drug development, elucidate disease mechanisms with greater fidelity, and ultimately accelerate the delivery of novel therapies to patients with cardiovascular disease.
Conclusion
hiPSC-CMs have firmly established themselves as an indispensable tool for modeling cardiovascular diseases, offering an unprecedented window into human-specific cardiac pathophysiology and drug responses. The integration of patient-specific genetics, advanced 3D culture systems, and gene editing technologies is progressively overcoming initial hurdles related to cellular immaturity and reproducibility. As the field moves forward, the convergence of AI-guided manufacturing, standardized maturation protocols, and the establishment of HLA-matched iPSC banks will be critical for scaling these platforms for widespread clinical and pharmaceutical application. The ongoing transition from research tool to a core component of drug discovery and personalized treatment pipelines heralds a new era in cardiovascular medicine, promising more effective, safer, and individually tailored therapies for patients worldwide.
References
- 1. Induced Pluripotent Stem Cell (iPSC) Industry Trends for ... [https://bioinformant.com/ipsc-industry-trends/]
- 2. Yamanaka factors and their importance in aging research [https://longevity.technology/yamanaka-factors/]
- 3. Human induced pluripotent stem cell-derived ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1687840/full]
- 4. Application of the Yamanaka Transcription Factors Oct4, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10531188/]
- 5. Yamanaka factors critically regulate the developmental ... [https://www.nature.com/articles/cr2008309]
- 6. Pluripotent Stem Cells: Recent Advances and Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12025069/]
- 7. Induced Pluripotent Stem Cell Reprogramming Protocols [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/cell-culture-and-cell-culture-analysis/stem-cell-culture/ipsc-reprogramming-protocols?srsltid=AfmBOorsH20yfVfW17odvxQXx50IwJR_WmZYia91oaoKtJmI2782s93c]
- 8. Development of a High-Efficacy Reprogramming Method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7555779/]
- 9. Induced Pluripotent Stem Cells (iPSC) and Their Use in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590075/]
- 10. Patient and Disease–Specific Induced Pluripotent Stem ... [https://www.sciencedirect.com/science/article/pii/S0031699724009451]
- 11. A comprehensive analysis of induced pluripotent stem cell ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1593207/full]
- 12. Induced pluripotent stem cells (iPSCs) [https://www.nature.com/articles/s41392-024-01809-0]
- 13. Advances in Induced Pluripotent Stem Cell Reprogramming ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577567/]
- 14. Efficient and reproducible generation of human iPSC ... [https://www.nature.com/articles/s41467-024-50224-0]
- 15. Human iPSC-derived cardiac-specific extracellular matrix ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12481509/]
- 16. hiPSC-derived cardiac fibroblasts dynamically enhance the ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1546483/full]
- 17. Delivery Methods for Generating iPSCs [https://blog.addgene.org/delivery-methods-for-generating-ipscs]
- 18. The emerging promise of induced pluripotent stem cells in ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925008461]
- 19. Transgene-Free Disease-Specific Induced Pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3652675/]
- 20. Progress in engineering functional cardiac tissues from ... [https://www.sciencedirect.com/science/article/abs/pii/S1742706125006762]
- 21. The molecular mechanisms of cardiac development and ... [https://www.nature.com/articles/s41392-024-02069-8]
- 22. Differentiation of Pluripotent Stem Cells for Disease Modeling [https://pmc.ncbi.nlm.nih.gov/articles/PMC10975450/]
- 23. Signaling Pathways Governing Cardiomyocyte Differentiation [https://pmc.ncbi.nlm.nih.gov/articles/PMC11202427/]
- 24. Endogenous Wnt/β-Catenin Signaling Is Required for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2886114/]
- 25. Endogenous Wnt/β-Catenin Signaling Is Required for Cardiac ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0011134]
- 26. Cardiac disease mechanobiology: advances using hiPSC-CMs [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515892/]
- 27. Human induced pluripotent stem cell-derived cardiomyocytes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571874/]
- 28. Clinical translation of human iPSC technologies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12498231/]
- 29. Induced Pluripotent Stem Cell (iPSC) Therapy Market Size, ... [https://www.precedenceresearch.com/induced-pluripotent-stem-cell-therapy-market]
- 30. A review of protocols for human iPSC culture, cardiac ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9405110/]
- 31. 5 Steps to Model Cardiac Disease [https://www.thermofisher.com/us/en/home/life-science/stem-cell-research/induced-pluripotent-stem-cells/cardiac-disease-modeling.html]
- 32. The role of iPSC research for insight into inherited ... [https://www.sciencedirect.com/science/article/pii/S1547527125025172]
- 33. The role of iPSC research for insight in inherited arrhythmia ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12554002/]
- 34. Induced Pluripotent Stem Cells for Cardiovascular Disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6708586/]
- 35. Arrhythmogenic Risk in iPSC-Derived Cardiomyocytes [https://www.mdpi.com/1648-9144/61/11/2056]
- 36. Human iPSC modeling of heart disease for drug ... [https://www.sciencedirect.com/science/article/pii/S245194562100101X]
- 37. Induced Pluripotent Stem Cells in Cardiomyopathy [https://pubmed.ncbi.nlm.nih.gov/40507801/]
- 38. Induced Pluripotent Stem Cells in Cardiomyopathy [https://www.mdpi.com/1422-0067/26/11/4984]
- 39. Preclinical Models of Cardiac Disease - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11116217/]
- 40. Modeling hypertrophic cardiomyopathy with human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9166443/]
- 41. Three-Dimensional iPSC-Based In Vitro Cardiac Models ... [https://www.mdpi.com/1422-0067/25/19/10690]
- 42. Experimental Models of Hypertrophic Cardiomyopathy [https://www.sciencedirect.com/science/article/pii/S2452302X24003966]
- 43. The use of artificial intelligence in induced pluripotent stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11108174/]
- 44. A new frontier in toxicity testing by evaluating drug-induced ... [https://pubmed.ncbi.nlm.nih.gov/40482844/]
- 45. Advanced cardiotoxicity profiling using field potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12646587/]
- 46. Predicting oncology drug-induced cardiotoxicity with donor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11199917/]
- 47. Early and non-destructive prediction of the differentiation ... [https://www.nature.com/articles/s41598-025-11108-5]
- 48. Induced pluripotent stem cells in disease modelling and drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584039/]
- 49. Human cardiac organoids: A recent revolution in disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10466470/]
- 50. 3D-bioprinting of patient-derived cardiac tissue models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10247285/]
- 51. A multi-cellular 3D bioprinting approach for vascularized ... [https://www.nature.com/articles/s41598-018-31848-x]
- 52. High-fidelity, personalized cardiac modeling via AI-driven ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523448/]
- 53. Rapid 3D BioPrinting of a human iPSC-derived cardiac ... [https://www.sciencedirect.com/science/article/pii/S2666102021000021]
- 54. Toward the latest advancements in cardiac regeneration using induced... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05499-8]
- 55. Frontiers | Integral approach to organelle profiling in human... [https://www.frontiersin.org/journals/toxicology/articles/10.3389/ftox.2025.1644119/full]
- 56. Cardiac disease mechanobiology: advances using hiPSC ... [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2025.1642931/full]
- 57. Induced pluripotent stem cells and personalized medicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC3254878/]
- 58. Engineering considerations of iPSC-based personalized ... [https://biomaterialsres.biomedcentral.com/articles/10.1186/s40824-023-00382-x]
- 59. Electrophysiological evaluation of human induced ... [https://www.sciencedirect.com/science/article/pii/S1873506121000222]
- 60. Disease modeling using human-induced pluripotent stem ... [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2022.966094/full]
- 61. Metabolic Maturation in hiPSC-Derived Cardiomyocytes [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389403/]
- 62. Maturation of human cardiomyocytes derived from induced ... [https://www.nature.com/articles/s41598-024-63905-z]
- 63. Structural and Functional Maturation of Cardiomyocytes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3699903/]
- 64. Current Advances and Future Directions of Pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11674482/]
- 65. Mind the gap: leak currents and induced pluripotent stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10445299/]
- 66. Metabolic Maturation Media Improve Physiological Function of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7437654/]
- 67. Simultaneous electro-dynamic stimulation accelerates ... [https://www.sciencedirect.com/science/article/pii/S0006291X24011410]
- 68. Electrical stimulation: a missing key to promote maturation ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1686342/full]
- 69. Generating 3D human cardiac constructs from pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8804169/]
- 70. Generation and maturation of human iPSC-derived 3D ... [https://www.nature.com/articles/s41598-022-22225-w]
- 71. Electrical Stimulation Promotes Cardiac Differentiation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4691644/]
- 72. Differentiation of Cardiomyocytes from Human Pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4447149/]
- 73. Engineering a controlled cardiac multilineage co ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12131582/]
- 74. Editorial: Role of induced pluripotent stem cells (iPSCs) in ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1734717/full]
- 75. Enhancing human pluripotent stem cell differentiation to ... [https://www.sciencedirect.com/science/article/pii/S2589004225007138]
- 76. Novel method of differentiating human induced pluripotent ... [https://www.nature.com/articles/s41598-023-31144-3]
- 77. The Promise and Challenge of Induced Pluripotent Stem Cells for... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5451899/]
- 78. Tumorigenicity risk of iPSCs in vivo: nip it in the bud - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9026204/]
- 79. Elimination of tumorigenic pluripotent stem cells from their ... [https://www.sciencedirect.com/science/article/pii/S221367112500147X]
- 80. Key tips and technologies for the development of iPSC- ... [https://www.biotechniques.com/drug-discovery-development/sartorius_ipsccgt_key-tips-and-technologies-for-the-development-of-ipsc-derived-cell-therapies/]
- 81. Tips and tricks for successfully culturing and adapting human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8640166/]
- 82. Building an expert system for quantitative monitoring of ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925005511]
- 83. Possible Strategies to Reduce the Tumorigenic Risk of ... [https://www.mdpi.com/1422-0067/25/10/5177]
- 84. CRISPR And iPSC Disease Modeling And Drug Screening [https://www.cellandgene.com/doc/crispr-and-ipsc-disease-modeling-and-drug-screening-0001]
- 85. CMN Weekly (10 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-10-october-2025-your-weekly-crispr-medicine-news/]
- 86. Artificial Intelligence for Drug Development [https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/artificial-intelligence-drug-development]
- 87. AI in Pharma and Biotech: Market Trends 2025 and Beyond [https://www.coherentsolutions.com/insights/artificial-intelligence-in-pharmaceuticals-and-biotechnology-current-trends-and-innovations]
- 88. The future of AI regulation in drug development: a comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12598624/]
- 89. CRISPR-Cas9 in Cardiovascular Medicine: Unlocking New ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763404/]
- 90. iPSC Summit 2025 | FujiFilm Cellular Dynamics, Inc. [https://www.fujifilmcdi.com/ipsc-summit-2025]
- 91. Human Pluripotent Stem Cell-Derived Cardiac Cells [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.655161/full]
- 92. Mixing experimental and computational models for a deeper ... [https://pubmed.ncbi.nlm.nih.gov/40741690/]
- 93. Maturation of human cardiac organoids enables complex ... [https://www.nature.com/articles/s44161-025-00669-3]
- 94. Patient-Specific iPSC-Based Disease Model for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5517015/]
- 95. Human-Induced Pluripotent Stem Cell Models for Amyloid ... [https://pubmed.ncbi.nlm.nih.gov/41295360/]
- 96. Modeling Cardiac Disease Mechanisms Using Induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7352327/]
- 97. Stem Cells Comparison to Human Cardiomyocytes [https://anabios.com/stem-cells-comparison/]
- 98. Induced Pluripotent Stem Cells for Cardiovascular ... Disease Modeling [https://pubmed.ncbi.nlm.nih.gov/29874173/]
- 99. Comparative analysis of the cardiomyocyte differentiation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9950567/]
- 100. Modeling human diseases with induced pluripotent stem cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC5868991/]
- 101. iPSC technology-Powerful hand for disease modeling and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4578564/]
- 102. iPSC-Derived Cardiomyocytes Taken to Rescue Infarcted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6127967/]
- 103. Automated inference of disease mechanisms in patient ... [https://www.nature.com/articles/s42003-025-08209-2]
- 104. From the Laboratory to the Clinic: Differentiation, Disease ... [https://pubmed.ncbi.nlm.nih.gov/41147803/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=0GaYCevddEDAfC5HA0wlElkFqaKqOUgRmbLrsDcYiaD&fc=None&ff=20251028144659&v=2.18.0.post22+67771e2]
- 105. iPSC-based Platforms Market 2025 Report and Trends [https://www.towardshealthcare.com/insights/ipsc-based-platforms-market-sizing]
- 106. The role of large language models in induced pluripotent ... [https://www.accscience.com/journal/JCTR/articles/online_first/5527]
- 107. Induced Pluripotent Stem Cells Market Size 2025 to 2034 [https://www.precedenceresearch.com/induced-pluripotent-stem-cells-market]
- 108. Trends & Strategies Shaping the $4.6+ Billion Induced ... [https://finance.yahoo.com/news/trends-strategies-shaping-4-6-145400149.html]
- 109. [Latest] Global Induced Pluripotent Stem Cells Production [https://www.globenewswire.com/news-release/2025/08/21/3137499/0/en/Latest-Global-Induced-Pluripotent-Stem-Cells-Production-Market-Size-Share-Worth-USD-4-34-Billion-by-2034-at-a-9-5-CAGR-Custom-Market-Insights-Analysis-Outlook-Leaders-Report-Trends.html]
- 110. Induced Pluripotent Stem Cells Market Size & Share Analysis [https://www.mordorintelligence.com/industry-reports/induced-pluripotent-stem-cells-market]
- 111. Induced Pluripotent Stem Cells Production Market 2025 [https://www.custommarketinsights.com/report/induced-pluripotent-stem-cells-production-market/]
- 112. Applications, challenges, and prospects of induced ... [https://www.sciencedirect.com/science/article/pii/S101684782400102X]
- 113. Engineering Heart Cells to Multiply - Stanford Medicine [https://med.stanford.edu/cvi/mission/news_center/articles_announcements/2025/engineering-heart-cells-to-multiply.html]
- 114. Improving cardiac differentiation of human pluripotent stem ... [https://www.sciencedirect.com/science/article/pii/S2352320424000191]