The Bone Marrow Niche: Decoding the Hematopoietic Stem Cell Microenvironment for Therapeutic Innovation
This article provides a comprehensive analysis of the hematopoietic stem cell (HSC) niche within the bone marrow microenvironment, a dynamic and complex regulatory unit essential for lifelong blood production.
The Bone Marrow Niche: Decoding the Hematopoietic Stem Cell Microenvironment for Therapeutic Innovation
Abstract
This article provides a comprehensive analysis of the hematopoietic stem cell (HSC) niche within the bone marrow microenvironment, a dynamic and complex regulatory unit essential for lifelong blood production. Tailored for researchers and drug development professionals, we explore the fundamental biology of niche components, from osteoblastic and vascular domains to perivascular stromal cells. The scope extends to cutting-edge methodologies for in vitro niche reconstruction, including 3D biomimetic models, organoids, and bone marrow-on-a-chip platforms. We further investigate the niche's role in disease pathogenesis, such as clonal hematopoiesis and myelodysplastic syndromes, and its emerging promise as a therapeutic target. Finally, we evaluate comparative and validation strategies that are refining our understanding of niche function in health and disease, offering a roadmap for translating basic science into clinical applications.
Deconstructing the HSC Niche: Cellular Architecture and Regulatory Mechanisms in Steady-State and Aging
The concept of the hematopoietic stem cell (HSC) niche, first proposed by R. Schofield in 1978, represents a foundational pillar in our understanding of stem cell biology [1]. Schofield's hypothesis postulated that a stem cell's fundamental capacity for self-renewal is intrinsically dependent on its association with a specific cellular environment, or "niche," that determines its behavior [1] [2]. For nearly five decades, this concept has driven scientific inquiry, evolving from a theoretical construct to a well-defined anatomical and functional unit within the bone marrow. Recent research has progressively refined this model, revealing a dynamic, multi-component regulatory system that governs HSC fate. Contemporary studies demonstrate that the niche is not merely a passive housing structure but an active participant in regulating the critical balance between HSC quiescence, self-renewal, and differentiation [3] [4]. This in-depth technical guide synthesizes historical perspectives with cutting-edge research, providing a comprehensive resource for scientists and drug development professionals engaged in hematopoietic stem cell niche bone marrow microenvironment research.
Historical Development and Conceptual Framework
The intellectual genesis of the niche concept lies in the earlier theory of the "hemopoietic-inductive microenvironment" (HIM), which posited that specific local environments instruct hematopoietic cell development [1]. Schofield's seminal contribution was to crystallize this idea into the "stem cell niche" hypothesis, providing a specific framework to explain the dependence of stem cells on their microenvironment for maintaining self-renewal capacity [1] [2]. This hypothesis was initially supported by observations that transplanted HSCs would only engraft when niche space was made available through conditioning regimens like irradiation [5].
Over time, two predominant interpretations of the niche have emerged, reflecting the complexity uncovered by experimental evidence. The table below summarizes this conceptual evolution.
Table 1: Conceptual Evolution of the Stem Cell Niche Hypothesis
| Aspect | Schofield's Original Postulate (1978) | Orthodox Interpretation | Alternative/Dynamic Interpretation |
|---|---|---|---|
| Core Definition | A cellular environment associating with stem cells to determine their behavior and self-renewal capacity [1]. | A confined anatomical site that supports self-renewal and maintains HSCs in a quiescent, undifferentiated state [1]. | A distinct, dynamic, hierarchical microenvironment regulating the balance between quiescence, proliferation, and differentiation of stem cells and progenitors [1]. |
| Primary Function | Maintain stem cell self-renewal and "stemness" [1]. | Retain HSCs in a quiescent state to protect the stem cell pool [1] [2]. | Instruct stem cell fate decisions dynamically in response to physiological demands and stressors [1] [4]. |
| Key Regulated Process | Self-renewal. | Quiescence maintenance. | Fate choice (quiescence, self-renewal, differentiation). |
The orthodox view emphasizes a static, protective role for the niche, primarily enforcing HSC quiescence. In contrast, the dynamic interpretation, supported by a growing body of evidence, recognizes the niche as a responsive entity that senses and reacts to changes such as injury, inflammation, and aging, thereby actively directing HSC fate [1] [4]. This evolution from a passive "space" to an active "instructional unit" marks a critical paradigm shift in the field.
Core Components and Regulatory Mechanisms of the HSC Niche
The bone marrow niche is a multicellular ensemble where diverse cell types coordinate to regulate HSCs. The major cellular constituents and their functional roles are detailed in the table below.
Table 2: Core Cellular Components of the Bone Marrow HSC Niche
| Cell Type | Key Identifiers/Markers | Primary Functions in Niche Regulation | Critical Secreted Factors |
|---|---|---|---|
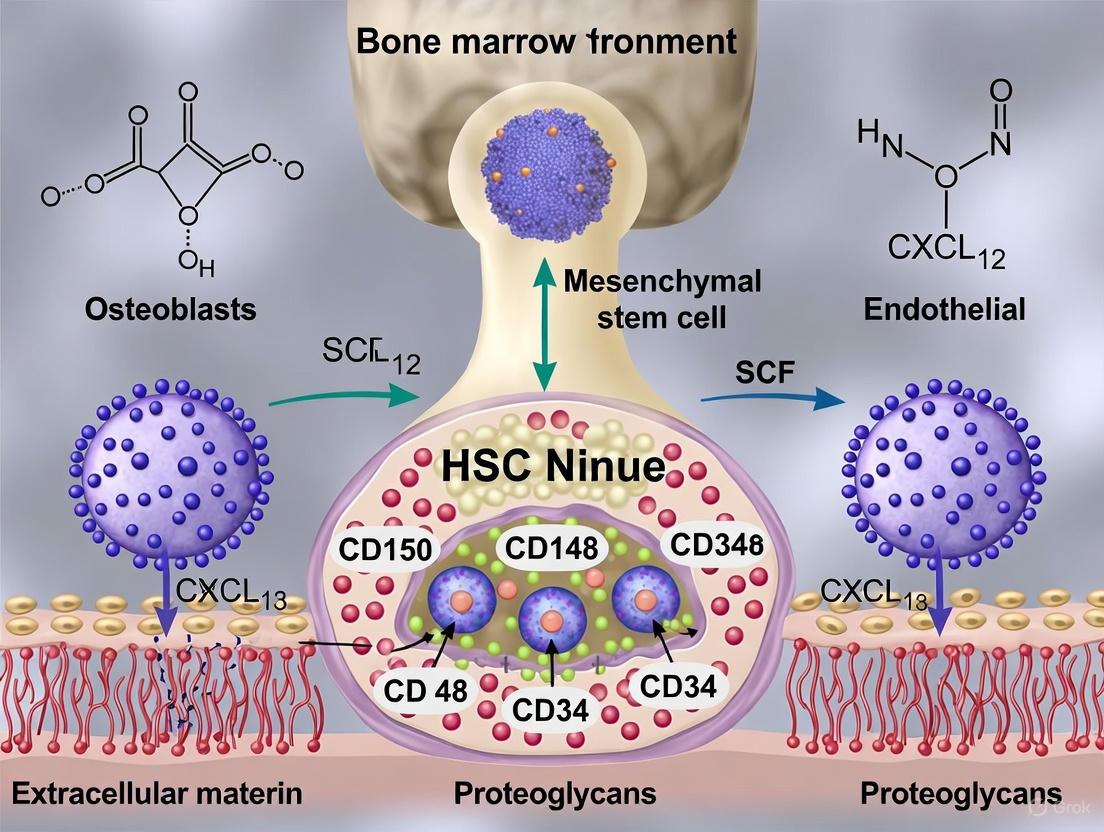
| Mesenchymal Stem/Progenitor Cells (MSPCs) | Nestin-GFP+, LEPR+, CXCL12-GFP+, CD51+CD140α+ [5] [6] [2] | Major source of CXCL12 and SCF; critical for HSC maintenance and retention; can differentiate into osteolineage cells [5] [6]. | CXCL12, SCF (Kitl), IL-7, Angiopoietin-1 [5] [6]. |
| Endothelial Cells (ECs) | CD31+, CD144+, SCA-1+ [5] [2] | Form vascular niches; regulate HSC quiescence and differentiation; facilitate homing and mobilization [3] [4]. | SCF, CXCL12, E-selectin [6] [2]. |
| Osteolineage Cells | Osteoblasts, osteocytes [2] | Historically considered a key niche component; contribute to endosteal niche; role in direct HSC maintenance is debated [6] [2]. | Osteopontin, Angiopoietin-1, Thrombopoietin [2]. |
| Sympathetic Nerves | N/A | Regulate HSC mobilization via circadian norepinephrine release; modulate CXCL12 expression; aid bone marrow regeneration [2]. | Norepinephrine [2]. |
| Megakaryocytes & Macrophages | N/A | Accessory niche cells; secrete factors that induce HSC quiescence [4]. | TGF-β, CXCL4 (PF-4) [4]. |
The signaling axis involving the C-X-C motif chemokine ligand 12 (CXCL12) and its receptor C-X-C motif chemokine receptor 4 (CXCR4) represents a cornerstone of niche regulation. This pathway is instrumental for HSC homing, retention, and quiescence [6]. CXCL12, produced predominantly by MSPCs and endothelial cells, acts as a potent chemoattractant for CXCR4-expressing HSCs. Beyond chemotaxis, CXCR4 signaling promotes HSC quiescence and facilitates access to other critical niche factors like Stem Cell Factor (SCF) [6]. The centrality of this pathway is highlighted by its additional role in guiding lymphoid progenitors to IL-7-producing niches for lymphopoiesis [6].
Diagram 1: Key Signaling in the HSC Niche.
Recent Experimental Advances and Technical Approaches
Challenging the Niche Size Dogma: Systemic and Local Regulation
The classical model posits that HSC numbers are directly limited by available niche space. However, a groundbreaking 2025 study using a novel femur-transplantation system challenges this view [5]. Researchers subcutaneously transplanted wild-type femoral bones into non-conditioned host mice, creating additional functional niches with intact MSPCs and vasculature but devoid of host HSCs.
Table 3: Key Findings from Femur-Transplantation Studies [5]
| Experimental Condition | Observation | Interpretation |
|---|---|---|
| Addition of 6 femoral grafts | Total body HSC numbers did not increase. | A systemic mechanism overrides local niche availability to limit total HSC numbers. |
| Transplanted femurs in hosts with defective endogenous niches | HSC numbers in grafts did not exceed physiological levels. | Local restraint also operates, preventing niche saturation even when HSCs are mobilized and available. |
| Role of Thrombopoietin | Thrombopoietin is pivotal in setting the total HSC number. | A specific systemic factor (Thrombopoietin) is a key determinant of the HSC set-point, independent of niche number. |
This research demonstrates that HSC numbers are subject to dual restriction—both systemically (body-wide) and locally (within the bone marrow)—and are not solely defined by niche capacity [5]. The identification of thrombopoietin as a key systemic regulator provides a molecular handle for this previously elusive mechanism.
Protocol: Femur Transplantation for Niche Studies
The following methodology was used to investigate niche regulation [5]:
- Graft Preparation: Femurs are harvested from donor adult mice (e.g., wild-type, nestin-GFP, or CD45.1 mice).
- Transplantation: Grafts are implanted subcutaneously into non-conditioned, non-irradiated host mice (e.g., WT or CD45.2 mice). Up to six femurs can be transplanted per host.
- Validation of Niche Viability: Grafts are analyzed at multiple time points (e.g., 1, 3, 5 months). Persistence of graft-derived MSPCs (CD45−TER-119−CD31−CD51+CD140α+) and host-derived endothelial cells is confirmed by flow cytometry and imaging. Host-derived haematopoietic cell repopulation is assessed.
- Functional Assessment: At endpoint, HSC numbers in host bones, grafts, and non-skeletal sites are quantified by flow cytometry (phenotype: Lin−SCA-1+KIT+CD150+CD48−CD34−). Niche function is tested via competitive bone marrow reconstitution assays.
Visualizing Niche Dynamics: Single-Cell Localization and Cycling
To move beyond snapshots and understand HSC behavior dynamically, researchers employ sophisticated genetic models. One powerful system is the hCD34tTA/Tet-O-H2BGFP transgenic mouse, which allows tracking of HSC division history [4]. In this model, HSCs express a histone H2B-GFP fusion protein. Upon administration of doxycycline (Doxy), new GFP synthesis is suppressed, and GFP intensity halves with each cell division, enabling the identification of HSCs that have divided 0 to 4+ times (G0 to GFP4) [4].
Using this model in aged mice, studies reveal niche-specific proliferation dynamics: the majority of HSCs surrounding arterioles retain high GFP signal (G0, dormant/slow-cycling), while HSCs associated with venules/sinusoids rapidly lose GFP label, indicating active cycling and a bias toward differentiation [4]. This demonstrates that different vascular niches instruct distinct HSC fates.
Diagram 2: HSC Division Tracking via a Genetic Model.
The Scientist's Toolkit: Key Research Reagent Solutions
Cutting-edge research into the HSC niche relies on a suite of specialized reagents, animal models, and methodologies.
Table 4: Essential Research Tools for HSC Niche Investigations
| Tool / Reagent | Function / Application | Key Examples / Models |
|---|---|---|
| Genetic Mouse Models | Lineage tracing, cell-specific ablation, and gene deletion in niche components. | Nestin-GFP [5] [2], Cxcl12-GFP [2], Lepr-Cre [6], Cdh5-creER (endothelial) [5], CD45.1/CD45.2 congenic [5]. |
| Cell Surface Markers for Isolation | Identification and purification of HSCs and niche cells by flow cytometry. | HSC (murine): Lin−SCA-1+KIT+CD150+CD48−CD34− [5]. MSPCs: CD45−TER-119−CD31−CD51+CD140α+ [5]. ECs: CD45−TER-119−CD31+ [5]. |
| In Vivo Functional Assays | Testing the long-term regenerative capacity and functional integrity of HSCs. | Competitive Bone Marrow Transplantation [5], Parabiosis [5]. |
| Mobilizing Agents | Studying HSC egress from the niche and the resulting compensatory mechanisms. | G-CSF [5]. |
| Advanced Microscopy & Lineage Tracing | Visualizing HSC location, division history, and niche interactions in real-time. | hCD34tTA/Tet-O-H2BGFP transgenic mice [4], multicolor confocal microscopy, intravital imaging. |
Fifty years after Schofield's prescient hypothesis, the field of HSC niche biology has matured from a theoretical model to a sophisticated understanding of a dynamic regulatory unit. The contemporary view defines the niche as a multi-tiered system employing local cellular cross-talk, systemic hormonal signals, and neural input to precisely control hematopoiesis. Recent discoveries of dual systemic/local HSC number control and niche-specific proliferation dynamics represent significant paradigm shifts [5] [4].
Despite these advances, the field faces challenges, including a lack of consensus on the precise definition and fundamental components of a "niche," which may be causing stagnation in conceptual progress [1]. Future research must leverage single-cell multi-omics and high-resolution spatiotemporal imaging to further deconstruct niche heterogeneity and plasticity. A major translational frontier lies in understanding how niches are corrupted in hematological malignancies and how to rebuild or modulate them for therapeutic benefit in regenerative medicine and stem cell transplantation. As we stand on the threshold of the 50th anniversary of Schofield's hypothesis, a concerted effort to integrate existing knowledge and standardize definitions will be crucial for the next generation of breakthroughs in this pivotal field [1].
The bone marrow microenvironment, or hematopoietic stem cell (HSC) niche, provides a specialized structural and functional unit essential for the maintenance, self-renewal, and differentiation of hematopoietic stem cells. The classical model distinguishes three principal niche compartments: the osteoblastic niche situated at the endosteal bone surface, the vascular niche comprising sinusoidal and arteriolar networks, and the perivascular niche where mesenchymal stromal cells create a supportive microenvironment for HSCs [7] [8] [9]. These niches are not isolated entities but form a highly integrated and dynamic system. Recent research has challenged the oversimplified dichotomy of endosteal versus vascular niches, revealing a more complex reality where these compartments are structurally and functionally intertwined, particularly during early myelopoiesis [7]. The precise coordination between these niches ensures lifelong hematopoiesis, while disruptions in their function contribute to hematological disorders and age-related hematopoietic decline. This technical guide provides a comprehensive analysis of these key cellular components, their regulatory mechanisms, and the experimental approaches used to study them, framed within the context of advanced bone marrow microenvironment research.
The Osteoblastic Niche
Anatomical Location and Cellular Composition
The osteoblastic niche, also termed the endosteal niche, is localized at the inner bone surface, in close proximity to the endosteum. This niche is predominantly composed of osteoblasts (bone-forming cells) and other bone-lining cells that create a specialized microenvironment for HSC regulation [8]. Osteoblasts anchor HSCs near the endosteal region and enhance hematopoiesis through the secretion of regulatory cytokines and adhesion molecules, thereby facilitating HSC homing and retention [10]. The functional significance of osteoblasts is evidenced by studies showing that osteoblast number correlates with HSC population size; ablation of osteoblasts leads to HSC reduction, while increased osteoblast numbers boost HSC quantities [10]. Beyond osteoblasts, this niche also contains osteoclasts (bone-resorbing cells) that regulate extracellular matrix turnover and release factors influencing HSC function and niche remodeling [8]. The coordinated activity of osteoblasts and osteoclasts maintains bone marrow integrity and composition, essential for proper hematopoietic function.
Regulatory Mechanisms and Signaling Pathways
Osteoblasts regulate HSC function through multiple mechanisms, including direct cell-cell contact and paracrine signaling. They secrete key regulatory factors such as osteopontin and angiopoietin-1 that help maintain HSC quiescence [8]. Additionally, osteoblasts guide HSC differentiation through Wnt and Bone Morphogenetic Protein (BMP) signaling pathways, thereby preserving hematopoietic regeneration capacity [8]. The angiopoietin-1/Tie2 receptor interaction is particularly crucial for maintaining HSC quiescence and adhesion within the niche [3]. Furthermore, osteoblasts produce thrombopoietin (TPO), a critical cytokine that promotes HSC maintenance and determines the total number of HSCs in the body, even in contexts of increased niche availability [5]. Recent evidence also highlights the role of osteoblast-derived SDF-1 (CXCL12) in preferentially regulating multipotent progenitors (MPP) and common lymphoid progenitors (CLP) retention [7].
Table 1: Key Signaling Molecules in the Osteoblastic Niche
| Signaling Molecule | Cellular Source | Function in HSC Regulation |
|---|---|---|
| Osteopontin | Osteoblasts | Regulates HSC quiescence and pool size |
| Angiopoietin-1 | Osteoblasts | Promotes HSC quiescence via Tie2 receptor interaction |
| Thrombopoietin (TPO) | Osteoblasts | Critical for HSC maintenance and determination of total HSC numbers |
| CXCL12 (SDF-1) | Osteolineage cells | Regulates retention of multipotent progenitors and lymphoid progenitors |
| Wnt Proteins | Osteoblasts | Guides HSC differentiation and self-renewal |
| BMP Signals | Osteoblasts | Influences HSC fate decisions |
Diagram 1: Osteoblastic niche signaling pathways regulating HSC fate.
The Vascular Niche
Sinusoidal and Arteriolar Compartments
The vascular niche encompasses the blood vessel networks within the bone marrow, primarily consisting of sinusoidal endothelial cells (SECs) and arteriolar endothelial cells (AECs). These two endothelial subtypes create distinct microenvironments that differentially regulate HSC function [10]. Sinusoidal vessels are characterized by their permeable, dilated structure and support HSC activation, trafficking, and mobilization into circulation [10]. In contrast, arteriolar vessels are surrounded by smooth muscle cells and non-myelinating Schwann cells, creating a niche that maintains HSC quiescence and protects against oxidative stress [10] [9]. The spatial distribution of HSCs within these vascular compartments correlates with functional states; HSCs in perisinusoidal areas are often more primed for differentiation and mobilization, while those associated with arterioles maintain greater quiescence [11]. This compartmentalization allows for precise regulation of hematopoietic output based on physiological demands.
Endothelial Cell Regulation of Hematopoiesis
Endothelial cells form the structural basis of the vascular niche and actively regulate HSC migration, maintenance, and activation through multiple mechanisms. They produce angiocrine factors—including VEGF, Notch ligands, and CXCL12—that directly influence HSC behavior [8]. The CXCL12/CXCR4 axis is particularly critical for HSC retention within the niche, with endothelial-derived SCF being specifically required for HSC maintenance and quiescence [7] [12]. Recent research has demonstrated that endothelial barrier integrity, controlled by FGF and CXCL12/CXCR4 signaling, is essential for HSC retention and metabolic stability [10]. Disruption of endothelial function leads to increased HSC mobilization, apoptosis, and reduced regenerative potential, highlighting the dynamic role of ECs in both stem cell maintenance and clinical mobilization strategies. The differential roles of endothelial subtypes are further emphasized by their distinct responses to stress and contribution to hematopoietic recovery.
Table 2: Functional Characteristics of Vascular Niche Components
| Vascular Component | Structural Features | HSC Functions Supported | Key Signaling Molecules |
|---|---|---|---|
| Sinusoidal Endothelial Cells (SECs) | Permeable, dilated structure with discontinuous basement membrane | HSC activation, trafficking, and mobilization; more permeable to circulating plasma | CXCL12, SCF, VEGF |
| Arteriolar Endothelial Cells (AECs) | Continuous basement membrane, surrounded by smooth muscle cells | HSC quiescence maintenance, protection from oxidative stress | Notch ligands, CXCL12, SCF |
| Type H Vessels | Specific endothelial subtype found in metaphysis | Association with osteogenesis, reduced in aged BM | Notch signaling components |
The Perivascular Niche
Mesenchymal Stromal Cell Populations
The perivascular niche represents a critical functional compartment where mesenchymal stromal cells (MSCs) create a supportive microenvironment for HSCs. Two principal MSC populations have been identified: Nestin-positive (Nestin⁺) MSCs and leptin receptor-positive (LepR⁺) MSCs, also known as CXCL12-abundant reticular (CAR) cells [12] [10] [9]. These perivascular stromal cells are strategically positioned around blood vessels and constitute the major cellular component of niches for HSCs and hematopoiesis in the bone marrow [9]. Nestin⁺ MSCs, predominantly located in perivascular regions in close association with sympathetic nerve fibers, are crucial for maintaining HSC quiescence and retention through the secretion of key factors such as CXCL12 and SCF [10] [9]. Conversely, LepR⁺ (CAR) cells constitute a significant portion of the adult BM stromal population and are instrumental in supporting hematopoiesis by contributing significantly to HSC homing, localization, and maintenance [10]. These cells also contribute to bone formation and adipogenesis, particularly during stress responses and following chemotherapy-induced damage.
Regulatory Functions of Perivascular Cells
Perivascular MSCs support hematopoiesis through multiple mechanisms, including the creation of a specialized extracellular matrix and provision of essential signaling cues. Through the secretion of key ECM proteins and cell adhesion molecules, MSCs regulate HSPC proliferation, differentiation, homing, retention, and maintain the quiescence necessary for effective hematopoiesis [10]. CAR/LepR⁺ cells have been demonstrated as the major cellular component of niches for HSCs, with approximately 97% of LT-HSCs in contact with these cells [9]. These perivascular cells produce critical niche factors including CXCL12, stem cell factor (SCF), VCAM-1, and Angpt1 [5]. The functional output of these signaling pathways is highly specific; for instance, although SCF is secreted by multiple stromal cells, HSC maintenance specifically relies on the endothelial source, demonstrating the precision of cellular crosstalk within the niche [7]. Furthermore, perivascular cells exhibit phenotypic and functional changes during aging, with increased expression of senescence markers like p16 and IL-1β, contributing to age-related hematopoietic decline [9].
Diagram 2: Perivascular MSC populations and their regulatory functions.
Integrated View of Niche Interactions
Cellular Crosstalk and Signaling Networks
The bone marrow niche operates as an integrated system where continuous crosstalk between cellular components ensures precise regulation of hematopoiesis. This complex multicellular communication involves not only the primary niche cells but also hematopoietic progeny and neural components that actively modulate niche function. Megakaryocytes, for example, regulate bone marrow hematopoiesis by secreting key cytokines such as TPO, CXCL4, and TGF-β to directly promote HSC maintenance, while also physically interacting with niche cells like osteomacs, osteoblasts, and osteoclasts to modulate their support of HSCs [7]. Similarly, sympathetic nerve fibers interact directly with MSCs, influencing the production of essential niche factors such as CXCL12, SCF, and VCAM-1, all critical for HSPC maintenance [10]. Recent research has uncovered novel regulatory mechanisms whereby dopamine, secreted by sympathetic nerves, promotes HSPC proliferation through upregulation of tyrosine-protein kinase Lck, which subsequently activates the MAPK pathway [10]. Additionally, regulatory T cells (Tregs) and cytotoxic T cells regulate the hematopoietic microenvironment via cytokines IL-10 and IFN-γ, respectively, and Tregs may engage in direct interactions with HSCs to establish a survival-promoting niche for aged HSCs [7].
Biophysical Properties of the Niche
Beyond biochemical signaling, the mechanical properties of cells, extracellular matrix (ECM), and tissues act as fundamental physical regulators within the bone marrow hematopoietic microenvironment. Key biophysical parameters such as stiffness, viscoelasticity, 3D topological architecture, and dynamic fluid shear stress critically regulate HSC quiescence, differentiation, migration, and apoptosis [7]. The ECM, secreted by niche cells, predominantly comprises structural proteins including type I and IV collagen, fibronectin, and laminin, alongside glycosaminoglycans such as hyaluronic acid and heparan sulfate proteoglycans [7] [12]. Matrix stiffness within the HSC niche is heterogeneous: the endosteal niche exhibits a relatively rigid matrix exceeding 35 kPa, whereas the vascular niche is characterized by softer matrices—approximately 0.3 kPa in bone marrow, 0.5–2 kPa in endothelium, and 5–8 kPa in vascular walls [7]. HSCs perceive ECM mechanical cues via mechanosensitive receptors, notably integrins, ion channels, and primary cilia, which collectively maintain functional homeostasis and precisely govern stem cell fate decisions [7]. Furthermore, specialized capillaries within the bone marrow regulate HSC survival via shear stress, with blood flow-induced shear stress activating specific signaling pathways in endothelial cells that influence HSC behavior.
Experimental Methodologies for Niche Analysis
Advanced Imaging and Modeling Techniques
Research on HSC niches employs sophisticated methodologies that enable high-resolution analysis of niche architecture and function. The iFAST3D imaging protocol allows high-resolution imaging of HSCs and their niche components within intact mouse tissues while preserving their spatial organization [11]. This technique involves sample preparation where bones are harvested and fixed, followed by shaving with a cryotome until the bone marrow is fully exposed to ensure optimal antibody penetration. After immunofluorescence staining with antibodies targeting HSC markers and niche components, imaging is performed using confocal laser scanning microscopy to capture z-stack images for 3D reconstruction and quantification of HSC size, shape, and spatial positioning relative to niche structures [11]. For in vitro modeling, 3D culture systems using biomimetic hydrogel scaffolds facilitate long-term HSC expansion and directed differentiation by establishing a three-dimensional niche architecture [7]. Microfluidic devices emulating vascular and osteogenic niches recreate the dynamic HSC microenvironment, offering platforms for in vitro modeling of hematological disorders and enabling high-throughput drug screening [7]. Additionally, bone transplantation models have been developed to rigorously define the role of niche size in regulating HSC numbers, enabling researchers to augment overall niche availability in vivo and assess the impact on HSC populations [5].
Molecular and Cellular Analysis
Single-cell technologies have revolutionized niche research by providing unprecedented resolution of cellular heterogeneity and molecular regulation. Single-cell RNA sequencing enables the identification of novel molecular regulators of HSC emergence and resolves cellular heterogeneity during hematopoietic development [13]. Flow cytometry-based size analysis determines the absolute size of individual HSPC populations from young and aged mice using forward scatter measurements calibrated with reference size beads [11]. To assess HSC polarity, the CellDetail analysis method involves immunofluorescence staining of FACS-isolated and fixed HSCs, followed by epifluorescence or confocal microscopy to capture subcellular high-resolution images of spatial distribution of proteins like Tubulin and Cdc42 within the cells [11]. For studying niche cell secretions, conditioned media collection from in vitro HSC niche models allows comparison of how young versus old niche environments affect hematopoietic cell development and function [14]. These complementary approaches provide comprehensive insights into niche function at molecular, cellular, and tissue levels.
Table 3: Essential Research Reagents and Experimental Tools for Niche Studies
| Research Tool | Application | Key Features/Components |
|---|---|---|
| iFAST3D Imaging | 3D spatial analysis of HSCs in intact bone marrow | Preserves spatial organization; uses antibodies against CD150, CD48, sinusoids, arterioles |
| Single-Cell RNA Sequencing | Molecular profiling of niche and HSC heterogeneity | Identifies novel regulators; resolves transitional cellular states |
| Conditioned Media from Niche Models | Analysis of secretory profiles from young vs. aged niches | Contains adipokines (e.g., adiponectin); reveals age-related inflammatory changes |
| Reference Size Beads | Flow cytometry-based HSC size measurement | Enables calibration with 7µm, 10µm, 16µm standards for precise sizing |
| Bone Transplantation System | In vivo niche expansion studies | Provides additional functional niches without adding HSCs |
| 3D Biomimetic Hydrogels | In vitro niche reconstruction | Replicates mechanical properties; supports long-term HSC culture |
The osteoblastic, vascular, and perivascular niches represent functionally distinct but highly integrated compartments that collectively regulate hematopoietic stem cell fate through complex biochemical and biophysical signals. Understanding the precise cellular composition, regulatory mechanisms, and dynamic interactions within these niches provides critical insights for both basic hematopoiesis research and clinical applications. Recent advancements in single-cell technologies, high-resolution imaging, and sophisticated in vitro models have progressively enhanced our understanding of niche biology, revealing unprecedented complexity in cellular crosstalk and microenvironmental regulation. These findings have important implications for developing novel therapeutic strategies for hematological disorders, improving hematopoietic stem cell transplantation outcomes, and addressing age-related hematopoietic decline. Future research focusing on the dynamic regulation of these niche components during homeostasis, stress, and aging will further advance our understanding of the bone marrow microenvironment and its role in health and disease.
Long-term hematopoietic stem cells (LT-HSCs) maintain lifelong blood production by residing in a state of quiescence within the hypoxic bone marrow niche. This in-depth technical guide explores the central role of hypoxia and the hypoxia-inducible factor-1α (HIF-1α) in enforcing a metabolic program essential for LT-HSC quiescence and functional preservation. We delineate how the hypoxic niche, through HIF-1α signaling, promotes a glycolytic metabolic phenotype and actively suppresses oxidative phosphorylation to minimize the production of reactive oxygen species (ROS), thereby protecting stem cell integrity. This review is framed within the broader context of bone marrow microenvironment research, synthesizing current molecular insights, experimental methodologies, and technical approaches relevant for scientists and drug development professionals working in hematopoiesis, stem cell biology, and regenerative medicine.
Hematopoietic stem cells (HSCs) are multipotent cells responsible for the lifelong regeneration of all blood cell lineages. The long-term self-renewing HSCs (LT-HSCs), which serve as the cornerstone of hematopoiesis, predominantly exist in a quiescent state (G0 phase of the cell cycle) within the bone marrow [15] [3]. This quiescence is a crucial mechanism to protect LT-HSCs from proliferative and genotoxic stress, thereby preserving their self-renewal capacity and preventing exhaustion [16] [15].
The bone marrow microenvironment, or "niche," is a complex, multicellular tissue where hematopoiesis occurs. A defining characteristic of this niche, particularly in the perisinusoidal regions where LT-HSCs are thought to reside, is its low oxygen tension (hypoxia) [15]. This hypoxic environment is not merely a passive condition but an active regulator of HSC function. Emerging evidence positions cellular metabolism as a fundamental determinant of HSC fate, with the hypoxic niche imparting specific metabolic characteristics that are integral to the maintenance of stemness [16] [15].
Metabolic Programming of Quiescent LT-HSCs
Quiescent LT-HSCs exhibit a distinct metabolic profile characterized by a reliance on anaerobic glycolysis for energy production, coupled with restrained mitochondrial activity. This bioenergetic configuration is a key adaptation to their hypoxic residence and is essential for their functional integrity.
Table 1: Metabolic Characteristics of Quiescent vs. Differentiating HSCs
| Metabolic Parameter | Quiescent LT-HSCs | Differentiating/Activated HSCs |
|---|---|---|
| Primary Energy Pathway | Anaerobic Glycolysis [16] [15] | Mitochondrial Oxidative Phosphorylation [16] |
| Mitochondrial Activity | Low membrane potential, inactive [16] [15] | High membrane potential, active [16] |
| Oxygen Consumption | Low [16] | High [16] |
| ROS Levels | Low (physiological) [16] [15] | Elevated [16] |
| ATP Levels | Lower [16] | Higher |
The preference for glycolysis over the more energy-efficient mitochondrial oxidative phosphorylation (OXPHOS) is a critical feature of stem cell maintenance. While OXPHOS generates more ATP per glucose molecule, it also produces reactive oxygen species (ROS) as byproducts. Elevated ROS levels can cause oxidative damage to DNA, proteins, and lipids, leading to impaired self-renewal capacity, loss of functionality, and accelerated HSC exhaustion [16] [15]. Therefore, the glycolytic metabolism of LT-HSCs serves to minimize ROS production, thus preserving genomic integrity and long-term regenerative potential [16]. This metabolic state, involving reduced oxidative capacity and lower mitochondrial activity, is considered a marker of "stemness" [16].
Central Role of HIF-1α in Metabolic Regulation
The transcription factor Hypoxia-Inducible Factor-1α (HIF-1α) is the master regulator mediating the adaptation of LT-HSCs to the hypoxic niche and enforcing their quiescent metabolic state.
Regulation of HIF-1α Stability
HIF-1α is highly elevated in LT-HSCs, partly through transcriptional regulation by Meis homeobox 1 (Meis1) [15]. The stability of the HIF-1α protein is exquisitely controlled by cellular oxygen levels:
- Under normoxic conditions: HIF-1α is continuously synthesized but rapidly ubiquitinated and degraded by the proteasome. This process is mediated by prolyl hydroxylase domain (PHD) enzymes, which hydroxylate HIF-1α in the presence of oxygen, marking it for recognition by the von Hippel-Lindau (VHL) E3 ubiquitin ligase complex [15].
- Under hypoxic conditions: PHD enzyme activity is inhibited, preventing HIF-1α hydroxylation. This stabilizes HIF-1α, allowing it to translocate to the nucleus, dimerize with its constitutive partner HIF-1β, and bind to Hypoxia-Response Elements (HREs) in the promoter regions of target genes [15].
HIF-1α Target Genes and Metabolic Control
Active HIF-1 orchestrates a metabolic switch by regulating a suite of genes that promote glycolysis and suppress mitochondrial OXPHOS.
- Promotion of Glycolysis: HIF-1 upregulates the expression of key glycolytic enzymes and glucose transporters, increasing the flux of glucose through the glycolytic pathway. This includes rate-limiting catalysts like phosphofructokinase-1 (Pfk-1) [15].
- Suppression of OXPHOS: A pivotal mechanism involves the HIF-1-mediated transactivation of genes encoding pyruvate dehydrogenase kinase (PDK1-4). PDK phosphorylates and inactivates the pyruvate dehydrogenase (PDH) enzyme. This critical step shunts glucose-derived pyruvate away from the mitochondria, preventing its conversion to acetyl-CoA and subsequent entry into the tricarboxylic acid (TCA) cycle. This effectively decouples glycolysis from mitochondrial respiration, limiting oxygen consumption and ROS generation [15].
The following diagram illustrates the core HIF-1α signaling pathway and its key metabolic functions in a hypoxic LT-HSC:
Experimental Models and Methodologies
Investigating the interplay between hypoxia, HIF-1α, and HSC biology requires a combination of genetic models, precise functional assays, and advanced analytical techniques.
Key Genetic Models and Functional Assays
Table 2: Experimental Models for Studying HSC Metabolism
| Model/Assay | Key Feature | Application & Functional Readout |
|---|---|---|
| HIF-1α Deletion/Modulation | Loss-of-function in HSCs or niche cells [15] [17]. | Assesses cell-autonomous vs. non-autonomous roles in HSC maintenance, quiescence, and metabolic programming. |
| P2H1Ad.Cortex Mouse Model | Adrenocortical-specific HIF1α deletion, causing chronic systemic elevation of glucocorticoids [17]. | Mimics chronic stress; used to study systemic hormonal influence on HSC quiescence and function. |
| Competitive Transplantation | Transplanting test HSCs (e.g., KO) with wild-type competitor cells into lethally irradiated recipients [15] [17]. | Gold-standard assay for evaluating long-term self-renewal and regenerative capacity in vivo. |
| Bone Marrow Niche Modeling (3D Cultures) | Co-culture of HSCs with stromal cells (MSCs) in hydrogels/scaffolds mimicking ECM [18]. | Enables dissection of cell-cell interactions and testing niche influences on HSC fate ex vivo. |
The experimental workflow for validating the role of a specific gene in HSC metabolism often follows a multi-step process, as visualized below:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Tools for HSC Metabolism Research
| Reagent / Tool | Function / Target | Application in HSC Research |
|---|---|---|
| Fluorescence-Activated Cell Sorter (FACS) | High-speed cell sorting and analysis. | Isolation of pure LT-HSC populations (e.g., Lin−Kit+Sca-1+CD48−CD150+) from murine bone marrow for functional and molecular analysis [15] [17]. |
| Antibody Panels for HSC Phenotyping | Surface markers: Lineage, c-Kit, Sca-1, CD48, CD150. | Identification and quantification of HSCs and progenitor subpopulations by flow cytometry [17]. |
| HIF-1α Stabilizers (e.g., PHD Inhibitors) | Chemical inhibition of PHD enzymes. | Experimentally mimic hypoxia and activate HIF-1α signaling in vitro to study downstream metabolic and functional effects [15]. |
| Seahorse Extracellular Flux Analyzer | Real-time measurement of OCR and ECAR. | Direct functional assessment of mitochondrial respiration (OXPHOS) and glycolytic flux in live HSCs [15]. |
| MitoTracker / MitoSOX Dyes | Fluorescent probes for mitochondrial mass and ROS. | Flow cytometry or microscopy-based quantification of mitochondrial content and superoxide production in HSCs [16] [15]. |
| Single-Cell RNA Sequencing (scRNA-seq) | Genome-wide expression profiling at single-cell resolution. | Unraveling HSC heterogeneity, identifying novel subpopulations, and mapping transcriptional states of niche cells [19] [20]. |
Pathophysiological Implications and Therapeutic Perspectives
Dysregulation of the finely tuned metabolic and signaling networks in LT-HSCs has significant consequences for disease pathogenesis and offers potential therapeutic avenues.
- Leukemogenesis: The metabolic dysregulation seen in leukemia often represents a corrupted version of stem cell metabolism. For instance, gain-of-function mutations in isocitrate dehydrogenase (IDH1/2) genes, found in acute myeloid leukemia (AML), lead to production of the oncometabolite 2-hydroxyglutarate (2-HG). This metabolite inhibits enzymes like TET2, leading to epigenetic alterations that block differentiation and promote leukemogenesis [16]. Furthermore, chronic inflammation, as studied in models of clonal hematopoiesis (CHIP) and myelodysplastic syndrome (MDS), can rewire the bone marrow niche. The emergence of inflammatory mesenchymal stromal cells (iMSCs) creates a self-reinforcing inflammatory loop that disrupts normal hematopoiesis and favors the expansion of mutant HSC clones, acting as a precursor to frank leukemia [19] [21].
- Aging and the Niche: The aged bone marrow microenvironment exhibits chronic, low-grade inflammation ("inflammaging"), characterized by elevated levels of cytokines like CCL5 and IL-1β [20]. This inflammatory state can impose metabolic stress on HSCs, driving a myeloid bias and potentially selecting for clones with pre-leukemic mutations, thereby linking niche aging to increased cancer risk [20] [21].
- Therapeutic Implications: Understanding HIF-1α-mediated metabolic regulation opens promising strategies. HIF stabilizers are being explored for their therapeutic potential [17]. Targeting the inflammatory bone marrow niche in pre-leukemic conditions like CHIP and MDS represents a novel preventive approach to intercept disease progression before leukemia develops [19] [21].
The maintenance of LT-HSC quiescence is a metabolically active process critically dependent on the hypoxic bone marrow niche and the transcriptional activity of HIF-1α. By enforcing a glycolytic metabolic state and suppressing mitochondrial OXPHOS, the HIF-1α pathway minimizes ROS production and protects the long-term self-renewal capacity of the stem cell pool. This mechanistic insight is fundamental to understanding both normal hematopoietic homeostasis and the pathophysiological processes of aging and malignant transformation. Future research, leveraging advanced genetic models, single-cell technologies, and sophisticated ex vivo niche systems, will continue to decipher the complex dialogue between HSCs and their microenvironment, paving the way for novel microenvironment-directed therapies for hematologic disorders.
The bone marrow microenvironment, or HSC niche, is a highly organized and dynamic structure essential for the lifelong maintenance of hematopoietic stem cells (HSCs). This niche provides precise regulatory signals that control HSC quiescence, self-renewal, and differentiation to maintain blood and immune system homeostasis [18]. During aging, this carefully tuned microenvironment undergoes profound functional and structural remodeling, which is now recognized as a critical driver of hematopoietic decline. The aged niche is characterized by a state of chronic, low-grade inflammation termed "inflammaging," which promotes a shift in HSC differentiation potential toward the myeloid lineage (myeloid bias) and impairs fundamental HSC functions [22] [23]. This transformation of the bone marrow landscape creates a self-reinforcing cycle that not only compromises normal hematopoiesis but also increases susceptibility to hematologic malignancies and other age-related pathologies [19]. Understanding the molecular and cellular mechanisms underlying niche aging is therefore paramount for developing therapeutic strategies to counteract age-related hematopoietic decline.
The Molecular and Cellular Hallmarks of the Aged Niche
Inflammaging and Senescence-Associated Secretory Phenotype (SASP)
Inflammaging describes the persistent, low-grade inflammation that characterizes the aging process. This phenomenon is driven by the accumulation of senescent cells within the bone marrow microenvironment. These cells exhibit a senescence-associated secretory phenotype (SASP), releasing a plethora of pro-inflammatory cytokines, chemokines, and growth factors [22] [23]. Key SASP factors include IL-6, IL-1β, TNF-α, and TGF-β, which create a chronically inflamed milieu that disrupts normal niche function [22] [23]. The production of these factors can be triggered by various sources of age-associated damage, including genomic instability, mitochondrial dysfunction, and oxidative stress [22]. This inflammatory signaling is not a passive consequence of aging but an active driver of hematopoietic dysfunction, establishing a vicious cycle where inflammation promotes further senescence and niche deterioration.
Table 1: Key Inflammatory Mediators in the Aged Bone Marrow Niche
| Mediator | Primary Cellular Source | Major Functional Impact on HSCs/Niche |
|---|---|---|
| IL-6 | Senescent stromal cells, Myeloid cells | Promotes myeloid-biased differentiation, Impairs self-renewal [23] |
| IL-1β | Myeloid cells, Senescent stromal cells | Expands pro-inflammatory neutrophil subsets, Drives HSC proliferation and myeloid skewing [20] |
| TNF-α | Immune cells, Senescent stromal cells | Contributes to chronic inflammatory signaling, Alters HSC differentiation potential [23] |
| TGF-β | Multiple niche cells | Associated with megakaryocytic differentiation bias; its neutralization can restore lymphoid potential [23] |
| Ccl5 (RANTES) | Aged microenvironment | Induces myeloid bias in young HSCs via mTOR pathway activation [20] |
Remodeling of the Cellular Niche
The functional decline of the aged HSC niche is driven by fundamental changes in its cellular composition. A critical transformation involves the replacement of normal, supportive mesenchymal stromal cells (MSCs) with inflammatory MSCs (iMSCs) [19]. These iMSCs secrete high levels of interferon-induced cytokines and chemokines, which recruit and activate T cells. These T cells then amplify the inflammatory signal, creating a feed-forward loop that sustains chronic inflammation, suppresses healthy hematopoiesis, and promotes vascular remodeling [19]. This process is evident in pre-malignant conditions like clonal hematopoiesis (CHIP), where inflammatory remodeling begins long before overt disease develops [19].
Concurrently, there is a documented expansion of megakaryocytes and megakaryocyte progenitors in the aged bone marrow [20]. While megakaryocytes are a normal component of the HSC niche and help regulate HSC quiescence via factors like CXCL4, their age-related expansion is thought to contribute to the dysregulation of HSC behavior, though the spatial relationship between HSCs and megakaryocytes during aging remains an active area of research [20]. The net effect of this cellular remodeling is the creation of a microenvironment that is inherently pro-inflammatory and selectively supportive of altered HSC function.
Myeloid Bias and Lineage Skewing
One of the most prominent functional consequences of an aged niche is the myeloid bias in HSC differentiation. Aging is associated with an increase in the proportion of myeloid-biased HSCs (my-HSCs) within the total HSC pool [24] [20]. This shift is driven by both cell-intrinsic changes in HSCs and powerful extrinsic pressures from the inflamed microenvironment. The molecular basis for this bias involves the epigenetic silencing of lymphoid-affiliated genes (e.g., EBF1, PAX5) and the upregulation of myeloid-specifying genes [25]. Furthermore, exposure of young HSCs to aged systemic factors or specific inflammatory cytokines like Ccl5 is sufficient to induce a myeloid-skewed output, demonstrating the instructive role of the extrinsic milieu [20]. This bias results in diminished lymphopoiesis—particularly a reduction in B cell production—and an expansion of myeloid cells, which contributes to weakened adaptive immunity and increased incidence of myeloid malignancies in the elderly [24] [20].
Functional Consequences of Niche Aging on Hematopoiesis
HSC Functional Decline
The aged bone marrow niche directly impairs the core functional properties of HSCs. Despite an overall increase in the phenotypic number of HSCs with age, these cells exhibit a reduced long-term self-renewal capacity and diminished functional competence in transplantation assays [24] [26]. This paradox—increased numbers but decreased quality—highlights the profound impact of aging on HSC biology. The aged niche contributes to this decline by disrupting the signals that normally maintain HSC quiescence. For instance, aged HSCs show impaired homing and engraftment capabilities, which is critical for successful bone marrow transplantation [26]. This homing defect is linked to alterations in the CXCL12/CXCR4 axis, a key pathway for HSC retention and maintenance within the niche [23]. The chronic inflammatory signaling also induces metabolic shifts in HSCs, forcing them to rely more on oxidative respiration than glycolysis, which in turn leads to the accumulation of reactive oxygen species (ROS) and increased DNA damage [23].
Table 2: Functional Alterations in Aged HSCs and Myeloid Cells
| Cell Type | Key Age-Related Functional Alterations | Underlying Mechanisms/Mediators |
|---|---|---|
| Hematopoietic Stem Cell (HSC) | Reduced self-renewal and engraftment potential [26] | Altered CXCL12/CXCR4 signaling, Accumulated DNA damage, Increased ROS [23] |
| Myeloid-biased differentiation [24] [27] | Epigenetic repression of lymphoid genes (e.g., EBF1, PAX5), Inflammatory cytokine signaling (e.g., IL-1, IL-6) [25] [23] | |
| Impaired homing to bone marrow [26] | Reduced response to homing signals, Altered adhesion molecule expression | |
| Neutrophil | Decreased phagocytic capacity, Abnormal chemotaxis [23] | Downregulation of CXCR2 receptor, Altered TLR function [23] |
| Increased pro-inflammatory subsets (IL-1β+) [20] | Exposure to aged bone marrow microenvironment | |
| Macrophage | Reduced efferocytosis (clearance of apoptotic cells) [20] | Diminished autophagy, Dysregulated cytokine secretion (e.g., reduced IL-10) [23] |
| Increased SASP factor secretion (e.g., IL-6, TNF-α) [23] | Accumulation of senescent cells, Chronic inflammatory signaling |
Immunosenescence and Inflammaging Loop
The functional decline of HSCs and the myeloid skewing of hematopoiesis directly lead to immunosenescence—the aging of the immune system. This is characterized by a shrinking pool of naïve T and B cells, a restricted T-cell receptor repertoire, and weakened responses to new antigens, resulting in reduced vaccine efficacy and increased susceptibility to infections [24] [20]. The aged niche fuels a vicious cycle: the immune cells it produces are dysfunctional and themselves contribute to inflammaging by secreting more pro-inflammatory cytokines (e.g., IL-1) [20]. This creates a self-reinforcing "inflammaging loop" where the inflammatory microenvironment produces defective immune cells, which in turn fail to resolve inflammation and adequately clear senescent cells, further exacerbating the inflammatory state of the niche [23] [20]. For example, aged macrophages show reduced phagocytic activity and efferocytosis, leading to the accumulation of cellular debris that further fuels inflammation [23].
Experimental Models and Methodologies for Studying the Aged Niche
Key Experimental Models
Heterochronic Transplantation
This gold-standard experiment involves transplanting HSCs from a donor of one age into a recipient of a different age, allowing researchers to disentangle the contributions of cell-intrinsic versus niche-extrinsic factors in HSC aging [20].
- Protocol:
- HSC Isolation: Harvest bone marrow from young (e.g., 2-3 month) and old (e.g., >20 month) mice. Enrich for HSCs using fluorescence-activated cell sorting (FACS) for the Lineage⁻Sca-1⁺c-Kit⁺ (LSK) CD150⁺CD48⁻ phenotype.
- Recipient Conditioning: Irradiate young and old recipient mice with a lethal dose to ablate endogenous hematopoiesis.
- Transplantation: Intravenously inject purified young HSCs into conditioned young and old recipients, and old HSCs into young and old recipients. Include competitor cells to fairly assess repopulation potential.
- Analysis: Monitor peripheral blood chimerism over 16+ weeks. Analyze lineage output (myeloid vs. lymphoid), conduct secondary transplants to assess self-renewal, and examine HSC homing and localization within the niche [20].
Single-Cell Multi-Omics Analysis
This approach provides an unbiased, high-resolution view of the cellular and molecular changes in the aged niche.
- Protocol:
- Sample Preparation: Obtain bone marrow from young and old donors (human or murine). Create a single-cell suspension.
- Single-Cell RNA Sequencing (scRNA-seq): Use a platform to capture thousands of individual cells and generate transcriptomic libraries. This identifies distinct cell populations (HSCs, MSCs, immune cells) and their inflammatory states [19] [27].
- Computational Analysis:
- Cluster Identification: Identify all cell types present in the bone marrow microenvironment.
- Differential Expression: Compare gene expression profiles between young and old cells to identify inflammaging signatures.
- Cell-Cell Communication: Infer altered signaling pathways between niche cells and HSCs [19].
- Spatial Validation: Validate findings using imaging techniques (e.g., immunofluorescence, imaging mass cytometry) on bone marrow biopsies to confirm changes in the spatial organization of the niche [19].
In Vitro Niche Reconstruction
Advanced 3D culture systems are being developed to model the complexity of the bone marrow niche in vitro for controlled studies of aging.
- Protocol:
- Scaffold Selection: Choose a biomaterial (e.g., gelatin-methacrylamide hydrogel, porous scaffolds) that mimics the physical and chemical properties of the native bone marrow extracellular matrix.
- Cellular Composition: Co-culture primary or induced pluripotent stem cell (iPSC)-derived HSCs with key niche cells, such as mesenchymal stromal cells (MSCs), endothelial cells, and osteoblasts.
- Inflammatory Priming: To model inflammaging, treat the culture with a cocktail of pro-inflammatory cytokines (e.g., IL-1β, TNF-α, IL-6).
- Functional Readouts:
- Assess HSC long-term culture-initiating cell (LTC-IC) capacity.
- Analyze differentiation bias by quantifying myeloid vs. lymphoid progeny.
- Measure transcriptomic and epigenetic changes in HSCs [28].
Diagram 1: The Inflammaging Signaling Cascade in the Aged Niche. This pathway illustrates how aging stressors trigger a self-reinforcing inflammatory loop that impairs HSC function.
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Research Tools for Studying the Aged HSC Niche
| Tool / Reagent | Primary Function in Research | Example Application |
|---|---|---|
| Cxcl5 Knockout (KO) Mice | To study the role of specific chemokines in niche-mediated lineage skewing. | Demonstrating that aged HSCs transplanted into Ccl5 KO recipients show reduced myeloid bias [20]. |
| SpliceUp Computational Tool | To identify cells with somatic mutations from single-cell RNA-seq data based on aberrant splicing. | Distinguishing mutant from non-mutant cells in clonal hematopoiesis (CHIP) and MDS samples to study niche interactions [19]. |
| CD49b Surface Marker | To prospectively isolate lymphoid-biased (CD49b+) and myeloid-biased (CD49b-) HSC subsets via FACS. | Investigating intrinsic age-related changes in functionally distinct HSC subpopulations [27]. |
| 3D Bone Marrow-on-a-Chip | To create a biomimetic in vitro model of the bone marrow niche with controlled fluid flow and cell-cell interactions. | Modeling the effects of chronic inflammatory cytokine exposure on HSC fate in a controlled system [28]. |
| Heterochronic Transplantation Model | To dissect cell-intrinsic (HSC) vs. extrinsic (niche) contributions to hematopoietic aging. | Transplanting young HSCs into aged recipients to test the rejuvenating effect of a young niche [20]. |
Diagram 2: Heterochronic Transplantation Experimental Workflow. This experimental design is fundamental for deconvoluting the intrinsic (HSC) and extrinsic (niche) contributions to hematopoietic aging.
The aged bone marrow niche is an active driver of hematopoietic decline, characterized by a self-perpetuating cycle of inflammation (inflammaging), cellular remodeling, and altered signaling that collectively promote myeloid bias and impair HSC function. Moving forward, research must focus on translating this mechanistic understanding into therapeutic strategies. Promising avenues include pharmacological inhibition of specific inflammatory pathways (e.g., IL-1 signaling), metabolic reprogramming of aged HSCs and niche cells, and epigenetic interventions to reverse myeloid-skewing gene expression programs [25] [26]. Furthermore, the development of increasingly sophisticated in vitro models, such as patient-specific bone marrow organoids, will enable high-throughput screening of rejuvenating compounds [28]. Ultimately, targeting the aged niche itself offers a powerful, complementary approach to direct HSC manipulation for preventing and treating age-related hematopoietic disorders and improving immune health in the elderly.
Clonal hematopoiesis of indeterminate potential (CHIP) is an age-related condition characterized by the expansion of hematopoietic stem cells (HSCs) bearing somatic mutations in genes associated with hematologic malignancies. While intrinsic driver mutations confer a selective advantage to HSCs, a growing body of evidence indicates that the bone marrow microenvironment, or HSC niche, plays a critical role in promoting the emergence and expansion of these pre-malignant clones. This review synthesizes current research on how age-related alterations in the HSC niche—including vascular dysfunction, increased adiposity, dampened DNA damage response, and pro-inflammatory signaling—create a permissive environment for clonal selection. We detail the molecular mechanisms underpinning this relationship and provide a comprehensive toolkit for researchers, including standardized experimental protocols, key reagent solutions, and visual schematics of critical pathways. Understanding these niche-driven dynamics is paramount for developing novel therapeutic strategies to mitigate the risk of malignant progression and associated morbidities in CHIP.
Clonal hematopoiesis (CH) describes a prevalent, age-associated state in which a genetically distinct subpopulation of blood cells, derived from a single hematopoietic stem or progenitor cell, expands within the bone marrow compartment [29] [30]. The defining criterion for Clonal Hematopoiesis of Indeterminate Potential (CHIP) is the presence of somatic mutations in genes recurrently mutated in hematologic malignancies at a variant allele frequency (VAF) of ≥2% in the absence of definitive diagnostic criteria for a hematologic neoplasm [29] [31]. The incidence of CHIP rises dramatically with age, affecting less than 1% of the population under 40 but 10-20% of individuals over 70 [30]. While often asymptomatic, CHIP confers an elevated, albeit low absolute, risk of progression to overt hematologic cancer and is strongly associated with increased all-cause mortality and non-malignant conditions, particularly cardiovascular disease [29] [32] [30].
The traditional understanding of CHIP has focused on the acquisition of driver mutations—such as in DNMT3A, TET2, ASXL1, and splicing factors—that confer a fitness advantage to HSCs, leading to their clonal expansion [29] [31]. However, the hematopoietic stem cell (HSC) niche, a complex microenvironmental network in the bone marrow, is now recognized as an equally critical determinant. With aging, this niche undergoes profound functional and phenotypic changes that can selectively support the expansion of mutant HSCs, thereby fueling pre-malignant progression [33] [34]. This review examines the mechanistic links between the aged HSC niche and CHIP, framing CH not merely as a cell-autonomous process but as a ecosystem-level phenomenon driven by a deteriorating microenvironment.
The Aged Hematopoietic Stem Cell Niche: A Permissive Environment
The HSC niche is a multi-component system essential for HSC maintenance, quiescence, and regenerative potential. Its age-related degeneration creates a permissive soil for the seeds of clonal hematopoiesis. Key alterations include:
- Cellular Composition Shifts: Aging is associated with a significant accumulation of bone marrow adipocytes [33] [34] [35]. This shift alters the secretory profile of the niche, increasing pro-inflammatory adipokines like adiponectin, which can dysregulate immune cell development and function [35]. Furthermore, the supportive osteolineage cells and mesenchymal stromal cells (MSCs) decline in function, impairing their ability to maintain HSC quiescence [33].
- Vascular Dysfunction: The vascular component of the niche, critical for HSC homeostasis, deteriorates with age. This manifests as increased vascular permeability (leakiness) and vessel dilation, driven by a loss of key regulatory factors like Netrin-1 from perivascular LepR+ cells [34]. This compromised integrity disrupts the hypoxic, protective milieu required for HSC dormancy.
- Altered Neural Regulation: The sympathetic nervous system, which innervates the niche to regulate HSC mobilization and maintenance, exhibits abnormal activity with age. Denervation or impaired nerve function can force HSCs out of their quiescent state and into active cell cycle, increasing their susceptibility to stress and potentially favoring clones resilient to such activation [33].
- Accumulation of DNA Damage: A fundamentally conserved attribute of aging niche cells (MSCs, endothelial cells) and HSCs is a dampened DNA damage response (DDR) and the accrual of DNA damage. This genomic instability creates a pro-mutagenic environment and compromises the functional potential of both the niche and the HSCs it supports [34].
Table 1: Hallmark Changes in the Aged HSC Niche and Their Functional Consequences
| Niche Component | Age-Associated Change | Functional Consequence |
|---|---|---|
| Mesenchymal Stromal Cells (MSCs) | Impaired function, increased adipogenic differentiation [34] | Reduced support for HSC quiescence, altered cytokine secretion (e.g., SCF, CXCL12) [33] |
| Vascular Endothelium | Increased permeability, vessel dilation [34] | Loss of HSC dormancy, altered HSC distribution [33] |
| Adipocytes | Expansion in bone marrow cavity [33] [35] | Secretion of pro-inflammatory adipokines (e.g., adiponectin), dysregulation of DC and HSC function [35] |
| Sympathetic Nerves | Abnormal nerve activity [33] | Disrupted HSC mobilization and maintenance of quiescence [33] |
| Global Niche | Attenuated DNA Damage Response (DDR) [34] | Accumulation of DNA damage in niche and HSCs, reduced regenerative capacity [34] |
Molecular Mechanisms Linking the Aged Niche to Clonal Expansion
The aged niche does not passively deteriorate; it actively selects for and promotes the growth of HSCs with specific mutations through several interconnected mechanisms.
Inflammation as a Central Driver
A bidirectional, self-reinforcing relationship exists between CHIP and inflammation. The aged niche is characterized by a state of chronic, low-grade inflammation ("inflammaging") [31] [35]. CHIP clones, particularly those with loss-of-function mutations in TET2 or DNMT3A, can contribute to this inflammation by producing macrophages and other immune cells that secrete elevated levels of pro-inflammatory cytokines such as IL-6, IL-8, and TNFα [31]. In a vicious cycle, this inflammatory milieu then selectively promotes the proliferation and survival of the mutant HSCs over their wild-type counterparts. For instance, HSCs with Tet2 or Dnmt3a mutations proliferate disproportionately when exposed to inflammatory signals like LPS or TNFα, leading to clonal expansion [29] [31].
Figure 1: The Vicious Cycle of Inflammation and Clonal Expansion. Inflammatory signals from the aged niche select for and promote the expansion of CH clones, whose mutant immune progeny in turn secrete more inflammatory cytokines, reinforcing the cycle and driving end-organ disease [29] [31].
Dysregulated Stem Cell-Niche Attachment
A mathematical modeling approach has illuminated how interactions with the niche directly influence clonal fitness [36]. This model posits that HSCs attached to the niche are quiescent. Upon detachment, they become activated and can either divide or differentiate. The progeny from division must reattach to the niche to maintain stemness. In this framework, the attachment and detachment rates are critical parameters determining a clone's long-term persistence. An aged niche, with its altered expression of adhesion molecules and chemokines (e.g., CXCL12), may disproportionately favor the attachment and retention of mutant clones, giving them a competitive edge over wild-type HSCs [36]. The model further suggests that the abundance of a clone in peripheral blood may not reflect its abundance in the niche, highlighting the diagnostic limitation of relying solely on blood samples [36].
Netrin-1 and DNA Damage Resolution
Recent research has identified Netrin-1 (NTN1) as a critical factor secreted by niche cells (MSCs and endothelial cells) that maintains niche and HSC fitness [34]. NTN1 plays an essential role in sustaining an active DNA damage response. With age, a decline in niche-derived NTN1 leads to the accumulation of DNA damage in both niche cells and HSCs. Remarkably, supplementation of aged mice with recombinant Netrin-1 was sufficient to rejuvenate the aged BM vascular niche, resolve accrued DNA damage, and restore the competitive fitness of aged HSCs to youthful levels [34]. This demonstrates that targeting specific deficiencies in the aged niche can directly counteract the functional decline that permits pre-malignant expansion.
Figure 2: Netrin-1 Mediated Niche Rejuvenation. Age-related decline in niche-derived Netrin-1 leads to a dampened DNA Damage Response (DDR) and accrued DNA damage, creating a permissive environment for CH. Exogenous NTN1 supplementation reactivates the DDR, restoring niche and HSC function [34].
Experimental Models and Methodologies for Niche-CHIP Investigation
In Vitro Modeling of the Aged HSC Niche
Objective: To simulate the secretory profile of young and aged HSC niches to study their effects on hematopoietic cell differentiation and function [35].
Protocol:
- Isolate Bone Marrow (BM) Cells: Harvest BM from the femurs, tibiae, humeri, ilia, and vertebrae of young (e.g., 2-3 months) and aged (e.g., 24 months) mice.
- Establish Long-Term Cultures: Resuspend viable BM cells in complete long-term culture medium (LTCM, e.g., MyeloCult M5300 with hydrocortisone). Seed cells at a density of 1.1 x 10^6 cells/cm² in culture flasks.
- Maintain Cultures: Culture cells for 4 weeks at 33°C with 5% CO2. Replace half of the medium with fresh LTCM weekly, taking care not to disturb the adherent layer.
- Generate Conditioned Media (CM): After 4 weeks, discard all LTCM, wash adherent cells with PBS, and add fresh supernatant media (e.g., RPMI-1640 with 10% FBS). After 48 hours, collect the CM.
- Process CM: Centrifuge CM at 1000 x g for 10 minutes at 4°C to remove cellular debris. Aliquot and store at -80°C. Pooling CM from multiple mice per age group is common to obtain sufficient volume and capture general trends [35].
- Functional Assays: Use the young and aged niche CM to differentiate BM-derived dendritic cells (BMDCs) or other hematopoietic progenitors. Analyze outcomes including:
- Cell Phenotype: Flow cytometry for maturation markers (e.g., MHC Class II, CD86).
- Functional Capacity: Mixed lymphocyte reaction (MLR) to assess T-cell stimulatory capacity.
- Cytokine Secretion: ELISA or multiplex assays to quantify secreted factors (e.g., IL-6) [35].
Genetic Mouse Models for Niche-Specific Manipulation
Objective: To delineate the role of specific niche-derived factors in HSC function and clonal hematopoiesis in vivo.
Protocol (Example: Netrin-1):
- Model Generation:
- Niche Phenotyping: Analyze the bone marrow of knockout and control littermates for:
- Hematopoietic Assessment:
- Flow Cytometry: Quantify frequencies of HSCs (Lin−cKIT+SCA1+CD150+CD48−) and multipotent progenitors (MPPs) in the BM [34].
- Functional Assays: Perform competitive repopulation assays to test the long-term engraftment and lineage output of HSCs from mutant niches [34].
- DNA Damage Analysis: Immunofluorescence for γH2AX or other DNA damage markers in HSCs and niche cells [34].
Mathematical Modeling of Clonal Dynamics
Objective: To quantify the impact of niche-mediated processes (attachment, detachment, proliferation) on the clonal composition of the stem cell compartment [36].
Framework:
- A published model uses a system of ordinary differential equations to track quiescent (niche-bound), active, and inactive HSC populations, alongside empty niche spaces [36].
- Key Parameters: Clone fitness is determined by its specific set of parameters: detachment rate, attachment rate, proliferation rate, and differentiation rate [36].
- Simulation: The model can be parameterized with data from mouse experiments and used to simulate clinical scenarios like bone marrow transplantation, clonal competition, and response to therapy. It can predict whether a clone will expand, go extinct, or coexist with others based on its interaction parameters with the niche [36].
Table 2: Key Quantitative Associations in CHIP and the Aged Niche
| Parameter | Observation/Measurement | Experimental/Clinical Context | Source |
|---|---|---|---|
| CHIP Prevalence | <1% (age <40) → 10-20% (age >70) | Analysis of large-scale human sequencing studies | [30] |
| Mortality Hazard Ratio | ~1.4 (all-cause mortality) | Meta-analysis of individuals with CHIP | [29] |
| Inflammatory Cytokines | Elevated IL-6, IL-8, TNFα | Plasma levels in individuals with CHIP, particularly with TET2 and DNMT3A mutations | [31] |
| Netrin-1 Rejuvenation | Restores competitive fitness of aged HSCs | In vivo treatment of aged mice with recombinant Netrin-1 | [34] |
| Clonal Abundance | Discrepancy between niche and blood | Predictions from mathematical modeling of HSC-niche interactions | [36] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Investigating the Niche-CHIP Axis
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Conditional Knockout Mice (e.g., LepR-Cre, Cdh5-CreERT2) | Enables cell-type-specific gene deletion in niche cells (MSCs, ECs). | Studying the role of niche-specific factors like Netrin-1 [34]. |
| Recombinant Netrin-1 | Recombinant protein for supplementation studies in vitro and in vivo. | Testing niche rejuvenation strategies in aged mouse models [34]. |
| MyeloCult M5300 Medium | Specialized medium for long-term culture of hematopoietic cells, supporting the growth of niche-forming stromal cells. | Generating in vitro models of the young and old HSC niche [35]. |
| Flow Cytometry Antibody Panels (e.g., Lin−, c-Kit+, Sca-1+, CD150+, CD48−) | Identification and isolation of pure populations of HSCs and progenitors by fluorescence-activated cell sorting (FACS). | Quantifying HSC frequencies and analyzing cell surface phenotype in different niche conditions [34]. |
| Digital Droplet PCR (ddPCR) | Ultra-sensitive and absolute quantification of low-frequency somatic mutations. | Detecting and tracking CHIP clones at very low Variant Allele Frequencies (<0.1%) [30]. |
| Cytokine Profiling Arrays | Multiplexed measurement of inflammatory cytokines (IL-6, TNFα, etc.) in conditioned media or serum. | Characterizing the inflammatory secretome of young vs. aged niche models or CHIP subjects [31] [35]. |
The pre-malignant expansion of HSCs in CHIP is a paradigm of how age-related ecosystem failure drives disease. The aged HSC niche is not a neutral bystander but an active accomplice, fostering clonal hematopoiesis through inflammatory signaling, dysfunctional cell-adhesion dynamics, and failure to maintain genomic integrity. The mechanistic insights linking niche-derived factors like Netrin-1 to DNA damage resolution offer a promising therapeutic avenue.
Future research must focus on translating these pre-clinical findings. This includes:
- Developing humanized mouse models to validate niche pathways in a human context.
- Conducting longitudinal clinical studies to correlate niche imaging parameters (e.g., BM adiposity) with CHIP clone dynamics.
- Launching clinical trials testing niche-directed therapies, such as anti-inflammatory agents (e.g., NLRP3 inflammasome inhibitors) or factors like Netrin-1, to suppress clonal expansion or mitigate its non-hematological consequences. By shifting the therapeutic focus from the mutant HSC alone to the dysfunctional niche that sustains it, we open a new frontier for preventing the progression of CHIP to malignancy and related morbidities.
Engineering the Niche: Advanced Models for Disease Modeling, Drug Screening, and Regenerative Therapy
The bone marrow (BM) microenvironment, or niche, is a complex physiological system indispensable for the maintenance, self-renewal, and differentiation of hematopoietic stem cells (HSCs). This niche not only supports hematopoietic homeostasis but also plays a significant role in the etiology of various hematological disorders [7]. For decades, research and drug development for blood cancers and disorders have relied on conventional two-dimensional (2D) culture systems and animal models. However, 2D cultures fail to replicate the spatial, metabolic, and cellular complexity of human bone marrow, while animal models are limited by significant species-specific differences in cellular composition, signaling molecules, and markers of the hematopoietic microenvironment [7] [37]. This lack of physiologically relevant models has hindered the development of effective treatments and accurate prediction of drug efficacy and toxicity.
The transition to three-dimensional (3D) biomimetic models represents a paradigm shift in hematopoietic research. These advanced systems aim to faithfully recapitulate the native BM architecture, incorporating critical elements such as stromal and vascular networks, hypoxic gradients, and extracellular matrix (ECM) components. Framed within the broader context of HSC niche research, these models are revolutionizing our fundamental understanding of hematopoiesis, disease pathogenesis, and the development of novel cell-based therapies [7] [38]. This whitepaper provides an in-depth technical guide to the core principles, methodologies, and applications of these transformative technologies.
Core Components of the Native and Engineered HSC Niche
Physiological Anatomy and Key Niches
In vivo, the HSC niche is a highly complex and dynamically regulated microenvironment. It is traditionally categorized into two primary, yet integrated, compartments:
- The endosteal niche, located near the bone surface, is rich in osteoblasts and osteolineage cells. It is characterized by a relatively stiffer matrix (exceeding 35 kPa) and is believed to host and maintain quiescent long-term HSCs (LT-HSCs) [7] [39].
- The vascular niche, associated with sinusoidal blood vessels and arterioles, comprises endothelial cells, perivascular stromal cells, and adipocytes. This region features a softer matrix (approximately 0.3 kPa in the marrow) and supports more active HSCs and progenitors [7] [40].
Emerging evidence indicates that these niches are highly integrated in both structure and function. The endosteal region is richly vascularized, forming a functional "arterial–endosteal niche" critical for early myelopoiesis [7]. Beyond cellular and biochemical components, the biophysical properties of the niche—including stiffness, viscoelasticity, 3D topology, and fluid shear stress—are fundamental physical regulators of HSC fate [7].
Critical Biochemical and Biophysical Cues
Successful in vitro reconstruction of the HSC niche requires the integration of specific biochemical and biophysical signals.
Table 1: Key Cues for Reconstructing the HSC Niche In Vitro
| Cue Category | Specific Factor/Property | Function in HSC Regulation |
|---|---|---|
| Biochemical Signals | CXCL12 (SDF-1)/CXCR4 axis | Guides HSC homing, retention, and survival [7] [40]. |
| Stem Cell Factor (SCF) | Critical for HSC maintenance and quiescence; specifically requires endothelial source [7]. | |
| VLA-4/VCAM-1 interaction | Mediates firm adhesion of HSCs to the niche, facilitating extravasation and lodging [40]. | |
| Biophysical Properties | Matrix Stiffness | Heterogeneous (0.3 - 40 kPa); directs HSC lineage commitment (e.g., softer matrices promote erythroid differentiation) [7] [41]. |
| Matrix Viscoelasticity | Influences HSC stemness and lineage-specific differentiation via mechanotransduction [7]. | |
| Oxygen Tension (pO₂) | Hypoxic conditions in specific zones help maintain HSC quiescence and stemness [39] [41]. | |
| Fluid Shear Stress | Activates signaling pathways in endothelial cells and HSCs, influencing survival and homing [7]. | |
| ECM Architecture | Fibrillar Fibronectin | Prevalent in the endosteal niche; presents growth factors and supports adhesion [39]. |
| Collagen I & IV, Laminin | Provide structural integrity and topological cues; distribution varies between niche sub-compartments [41]. |
Figure 1: Key Signaling Pathways and Microenvironmental Cues Regulating HSC Fate. The diagram illustrates how biochemical signals, biophysical cues, and ECM architecture from the bone marrow niche collectively regulate critical HSC functions like homing, quiescence, and differentiation.
Leading-Edge 3D Biomimetic Platforms and Methodologies
Biomimetic Scaffolds and Hydrogel Systems
A primary approach involves using 3D scaffolds, particularly hydrogels, to mimic the native ECM. These systems are functionalized with specific physical and biochemical cues to direct HSC behavior.
- Soft Collagen-I Hydrogels for LT-HSC Maintenance: A landmark study demonstrated that soft collagen type-I hydrogels drive nestin and HIF-1α expression in perivascular stromal cells (PerSCs). When CD34+ HSCs were co-cultured in these bioengineered niches, LT-HSC numbers were maintained with normal clonal and in vivo reconstitution potential, without media supplementation [39]. The system also supported the survival of CRISPR-edited HSCs, highlighting its clinical potential [39].
- Bioemulsions for Architectural Mimicry: To capture the dense, adipocyte-rich architecture of the BM with its anisotropic mechanical properties, researchers have used bioemulsions. Here, oil microdroplets recreate the architectural features of the hematopoietic niche. Mesenchymal stem cells (MSCs) grown at the surface of these bioemulsions assemble an interstitial matrix and secrete critical factors. This platform achieved a significant (>33-fold) expansion of HSCs compared to suspension cultures and enabled scale-up in bioreactors [42].
- Polymer-Controlled Fibronectin Presentation: Surfaces like poly(ethyl acrylate) (PEA) can be used to control the conformation of adsorbed fibronectin, driving it into a physiological fibrillar structure. This unfolding exposes key integrin-binding (RGD) and growth-factor-binding (P5F3) domains, enhancing GF presentation and signaling to create a more authentic endosteal niche-like environment [39].
Table 2: Quantitative Performance of Advanced 3D Biomimetic Platforms
| Platform Type | Key Functional Components | Reported HSC Expansion / Maintenance | Key Advantage |
|---|---|---|---|
| Soft Collagen-I Hydrogel [39] | Nestin+/HIF-1α+ PerSCs, Fibrillar ECM | Maintenance of LT-HSCs with normal in vivo reconstitution potential. | Maintains naive LT-HSC pool without cytokine supplementation. |
| Bioemulsion Platform [42] | Oil microdroplets, MSC-derived matrix, Mechanical anisotropy | >33-fold expansion compared to suspension cultures. | Enables scalable expansion in flask bioreactors (2M cell batches). |
| 3D Bone Marrow Niche (Crown Bioscience) [43] [44] | Biofunctional hydrogel, Stromal & Endothelial cells | Enables cancer cell proliferation & drug testing for up to 7 days. | Recapitulates cell adhesion-mediated drug resistance (CAM-DR) for predictive screening. |
Bone Marrow Organoids and Organ-on-a-Chip Systems
Beyond scaffold-based approaches, more complex, self-organizing systems are emerging.
- Bone Marrow Organoids (BMOs): These are 3D constructs that aim to closely mimic the human BM microenvironment by incorporating multiple cell populations and structural elements [40]. While true, self-renewing hematopoietic organoids are still an unmet goal, current organoid-like systems are valuable for preclinical testing of gene-edited HSCs and for investigating human-specific HSC-niche interactions beyond animal models [40] [38]. Current challenges include incomplete tissue recapitulation, lack of vascularization, and batch variability [40].
- Microfluidic Niche-on-a-Chip Devices: These systems emulate the dynamic HSC microenvironment, including vascular perfusion and mechanical forces like shear stress. They offer platforms for in vitro modeling of hematological disorders and enable high-throughput drug screening [7] [38]. These devices allow for controlled delivery of pharmacologic agents under defined oxygen and nutrient gradients, closely emulating marrow sinusoids [38].
The Scientist's Toolkit: Key Research Reagent Solutions
The development of these sophisticated models relies on a suite of essential materials and reagents.
Table 3: Essential Research Reagents for 3D Bone Marrow Niche Modeling
| Reagent Category | Specific Examples | Function in Niche Reconstruction |
|---|---|---|
| Biomaterial Scaffolds | Collagen-I Hydrogels, Polyethylene Glycol (PEG)-based Hydrogels, Fibrin, Matrigel | Provide a 3D structural support that mimics the native ECM, allowing cell encapsulation and network formation [39] [38] [41]. |
| Soluble Factors & Cytokines | CXCL12 (SDF-1), Stem Cell Factor (SCF), Thrombopoietin (TPO), BMP-2 | Recreate critical biochemical gradients for HSC maintenance, homing, quiescence, and differentiation [7] [39] [40]. |
| Engineered Surfaces | Poly(ethyl acrylate) (PEA) | Controls the nanoscale organization of adhesive proteins like fibronectin, exposing key binding domains for enhanced cellular signaling [39]. |
| Cellular Components | Primary Mesenchymal Stromal Cells (MSCs), Perivascular Stromal Cells (PerSCs), Umbilical Cord Vein Endothelial Cells (HUVECs), Patient-derived tumor cells | Recreate the multicellular crosstalk of the niche. Stromal cells are often used to form feeder layers that support HSCs [42] [43] [39]. |
Detailed Experimental Protocol: Establishing a Biomimetic Hydrogel Co-culture
The following protocol outlines the key steps for creating a bioengineered niche using soft collagen hydrogels to maintain LT-HSCs, based on the methodology described in Nature Communications [39].
Figure 2: Experimental Workflow for Biomimetic Hydrogel Co-culture. This flowchart outlines the key steps for establishing a bioengineered niche capable of maintaining long-term hematopoietic stem cells (LT-HSCs).
Step-by-Step Protocol
Step 1: Hydrogel Fabrication
- Procedure: Prepare a solution of type-I collagen at a low concentration (e.g., 1.5 mg/mL) to achieve a soft matrix with a stiffness of <1 kPa. Neutralize the acidic collagen solution using NaOH and an appropriate buffer (e.g., 10x PBS) according to the manufacturer's instructions. Pipet the neutralized collagen into the desired culture vessels (e.g., multi-well plates) and incubate at 37°C for 30-60 minutes to induce polymerization, forming the 3D hydrogel.
Step 2: Stromal Cell Seeding
- Procedure: Isolate and expand human perivascular stromal cells (PerSCs) or bone marrow-derived mesenchymal stromal cells (MSCs) in standard 2D culture. Trypsinize the cells, resuspend in appropriate stromal medium (e.g., DMEM supplemented with FBS), and seed onto the surface of the polymerized collagen hydrogels at a density optimized for confluence (e.g., 50,000 cells/cm²).
Step 3: Stromal Conditioning
- Procedure: Culture the stromal cell-laden hydrogels for 5-7 days to allow the cells to condition the matrix and adopt a niche-supportive phenotype. The soft mechanical environment will drive the upregulation of nestin and HIF-1α in the stromal cells, which is critical for their HSC-supportive capacity. Verify phenotype induction via immunofluorescence staining or RT-qPCR for nestin and HIF-1α.
Step 4: HSC Introduction
- Procedure: Isolate CD34+ hematopoietic stem and progenitor cells (HSPCs) from a source such as umbilical cord blood or peripheral blood. Resuspend the CD34+ cells in a minimal, cytokine-free basal medium (e.g., IMDM with no added growth factors). Gently seed the HSCs onto the pre-conditioned stromal niche. Allow cells to settle and interact with the niche for several hours before any medium changes.
Step 5: Culture and Analysis
- Procedure: Maintain the co-culture for 7-14 days without cytokine supplementation. Change half of the medium with fresh basal medium every 3-4 days.
- Functional Analysis: To assess LT-HSC function, use the Long-Term Culture-Initiating Cell (LTC-IC) assay. After the culture period, recover the HSCs and plate them onto a secondary, irradiated stromal feeder layer. Maintain this secondary culture for 5+ weeks with weekly half-medium changes. The number and type of colonies formed (measured by a limiting dilution assay or cobblestone area-forming cell count) indicate the frequency of primitive LT-HSCs present in the primary 3D culture [39].
- In Vivo Validation: The gold standard for validating LT-HSC function is transplantation into immunodeficient mice (e.g., NSG mice). Engraftment and multi-lineage reconstitution assessed 3-6 months post-transplantation confirm the preservation of true LT-HSCs [39].
Applications in Research and Drug Development
The deployment of 3D biomimetic bone marrow models is transforming preclinical research in several key areas:
Disease Modeling and Drug Screening for Hematological Malignancies: 3D models like Crown Bioscience's Bone Marrow Niche (BMN) platform provide a physiologically relevant system for studying liquid malignancies such as acute myeloid leukemia (AML) and multiple myeloma. These models recapitulate critical mechanisms like cell adhesion-mediated drug resistance (CAM-DR), where cancer cells binding to matrix components trigger survival pathways that blunt therapy effects [43] [44]. This allows for more predictive evaluation of drug efficacy and identification of compounds that can overcome microenvironment-induced resistance [43] [37] [38].
Ex Vivo HSC Expansion for Transplantation: A primary clinical motivation is the expansion of HSCs for transplantation, which is currently constrained by poor cell availability. Platforms that significantly expand HSCs, such as the bioemulsion system (>33-fold) or those that maintain LT-HSCs, like the soft collagen hydrogel, are groundbreaking [42] [39]. Successful translation could alleviate the reliance on matched donors and improve outcomes for patients undergoing hematopoietic stem cell transplantation.
Toxicity Testing and Safety Pharmacology: The bone marrow is a primary site for dose-limiting toxicity for many chemotherapeutics and other drugs. 3D BM models are increasingly being used for hematopoietic toxicity testing, providing a more human-relevant platform to assess drug-induced effects on stem cell survival and function earlier in the drug development process, potentially reducing late-stage failures and animal testing [43] [45].
Gene Therapy and Editing: The ability to maintain and manipulate LT-HSCs ex vivo is crucial for advancing gene therapies. The bioengineered niche comprising nestin/HIF-1α expressing PerSCs has provided proof-of-concept for supporting the survival of CRISPR-edited HSCs, a critical step towards developing safer and more effective genetic treatments for blood disorders [39] [40].
The field of 3D biomimetic bone marrow modeling has progressed from simple stromal co-cultures to sophisticated, multi-parametric systems that integrate critical aspects of the native niche architecture, mechanics, and cellular composition. These models are now indispensable tools for deconstructing hematopoietic physiology and pathology, offering unprecedented translational potential.
Future developments will focus on increasing physiological fidelity and functionality. Key directions include the integration of functional vasculature through advanced bioprinting or self-assembly techniques, the creation of true self-renewing bone marrow organoids, and the incorporation of additional niche residents like osteoblasts, osteoclasts, and nervous system components [44] [38]. Furthermore, the integration of artificial intelligence (AI) and machine learning with high-content imaging and multi-omics data from these 3D models will deepen our understanding of the niche and accelerate predictive drug screening [7] [38].
As these technologies continue to mature and standardize, they are poised to bridge the critical gap between traditional 2D assays and clinical outcomes, ultimately accelerating the development of novel, effective therapies for hematological cancers and disorders.
The bone marrow (BM) microenvironment, or hematopoietic stem cell (HSC) niche, is a complex, multi-component system essential for the lifelong regulation of hematopoiesis. It consists of heterogeneous cell populations, signaling molecules, and extracellular matrix (ECM) proteins that collectively regulate HSC fate decisions including quiescence, self-renewal, and differentiation [46] [47]. Advancements in stem cell biology and microengineering have yielded two transformative platforms for modeling this niche: bone marrow organoids (BMOs) and microfluidic Bone Marrow-on-a-Chip systems. These technologies overcome the limitations of traditional 2D cultures and animal models by recapitulating the human BM's spatial, mechanical, and biochemical complexity, offering unprecedented opportunities for studying hematopoietic development, disease modeling, and preclinical drug safety profiling [48] [49] [50].
The HSC niche primarily exists in two specialized anatomical locations: the endosteal niche, localized at the bone surface and enriched with osteoblasts and mesenchymal stem cells that support HSC quiescence and self-renewal; and the perivascular niche, adjacent to BM vasculature and composed of endothelial cells and mesenchymal stromal cells that promote proliferation and differentiation [46] [47]. These niches are not merely cellular but incorporate critical biomechanical forces, oxygen tension gradients, and a dynamic ECM that provides structural support and regulates growth factor availability [47]. The development of physiologically relevant in vitro models requires faithfully integrating these multifaceted components, a challenge now being addressed through the platforms detailed in this technical guide.
Bone Marrow Organoids (BMOs) from Human Induced Pluripotent Stem Cells
Protocol for Generating Complex BMOs
A landmark 2024 Nature Methods study established a robust, serum-free protocol for generating complex BM-like organoids from human induced pluripotent stem cells (iPSCs) [49]. This method recapitulates key aspects of embryonic development to yield a self-organizing, 3D structure containing hematopoietic, mesenchymal, and endothelial components. The detailed, stepwise methodology is as follows:
- Day -3: Embryoid Body (EB) Formation. iPSCs are aggregated to form EBs, initiating spontaneous differentiation and germ layer specification.
- Day 0: Mesoderm Induction. EBs are treated with a cocktail containing the Wnt agonist CHIR99021, bone morphogenetic protein 4 (BMP4), and vascular endothelial growth factor (VEGF) to direct differentiation towards mesodermal lineages, the precursor to blood and vascular tissues.
- Day 2: Hemogenic Endothelium (HE) Induction. The activin/nodal pathway inhibitor SB431542 is added alongside basic fibroblast growth factor (bFGF), stem cell factor (SCF), and VEGF to pattern the mesoderm and induce the formation of hemogenic endothelium, a transient population that gives rise to hematopoietic cells.
- Day 4: Matrix Embedding for Self-Assembly. The patterned EBs are embedded into a collagen I/Matrigel matrix to provide a 3D scaffold that supports the self-organization of cells into complex tissue structures.
- Day 8: Vascular Enhancement. A low dose of VEGF is added to the culture to promote the formation and stabilization of vascular networks within the developing organoid.
- Day 10: Organoid Maturation. The sprouted EBs are transferred to an ultra-low-attachment 96-well plate to permit further maturation and spherical organoid formation without adhesion to plastic. The organoids are maintained without lineage-directing cytokines to assess intrinsic differentiation potential, typically maturing until at least day 17 [49].
This workflow results in spherical organoids with a mean diameter of approximately 1,300 µm that contain a heterocellular composition reflective of the native BM niche [49].
Key Characteristics and Validation of BMOs
Single-cell RNA sequencing and high-resolution imaging confirm that BMOs recapitulate critical structural and functional features of the BM microenvironment. The cellular composition is highly representative, as shown in the table below.
Table 1: Cellular Composition of iPSC-Derived Bone Marrow Organoids (at Day 17)
| Cell Type | Key Markers | Average Percentage per BMO | Functional Role in Niche |
|---|---|---|---|
| Hematopoietic Cells | CD45+ | 39.3% | Includes progenitors and mature myeloid cells |
| Mesenchymal Stromal Cells (MSCs) | CD45−CD31−CD271+ | 41.3% | Niche support cells; source of key factors like CXCL12 |
| Endothelial Cells (ECs) | CD45−CD31+ | 6.0% | Forms vascular networks |
| HSPCs | CD45+CD11b−CD34+ | 1.42% | Hematopoietic Stem and Progenitor Cells |
| Multipotent MSPCs | CD45−CD31−CD271+CD90+CD105+CD73+ | 0.96% | Mesenchymal Stem/Progenitor Cells with differentiation capacity |
Spatial analysis via confocal and two-photon microscopy reveals that BMOs self-organize into anatomically relevant structures, including a vessel-like network of CD31+ endothelial cells enveloped by PDGFRβ+ pericytes [49]. Critically, the organoids contain a reticular network of CXCL12-abundant reticular (CAR) cell-like cells and Nestin+ stromal cells extending protrusions toward the vasculature, mirroring the in vivo perivascular niche essential for HSC maintenance [49]. Functionally, BMOs support the presence of hematopoietic stem/progenitor cells (HSPCs) with a fetal HSC-like transcriptional signature, demonstrated lymphoid potential, and a subset even showed transient engraftment potential upon xenotransplantation into immunodeficient mice [49]. The platform's utility for disease modeling was validated using iPSCs from a patient with VPS45 deficiency, an inborn error of hematopoiesis, successfully recapitulating disease-specific phenotypes [49].
Microfluidic Bone Marrow-on-a-Chip Platforms
Design Principles and Experimental Workflow
Microfluidic Bone Marrow-on-a-Chip platforms leverage microscale engineering to create a perfused, dynamic model that incorporates essential physiological cues absent in static cultures, such as fluid shear stress and nutrient/waste gradients [48] [50]. A representative and advanced chip design, the HUMIMIC Chip2 system, utilizes a closed microfluidic circuit containing a porous zirconium oxide ceramic scaffold [50]. This scaffold provides a large surface area and 3D structure for co-culture, mimicking the spongy architecture of trabecular bone.
The standard protocol for establishing a functional bone marrow model within this chip is as follows:
- Step 1 - Stromal Preculture: Primary human bone marrow-derived mesenchymal stromal cells (MSCs) are expanded in 2D culture for 7-15 days and then seeded onto the ceramic scaffold. The scaffold is cultured under static conditions for 10 days, allowing the MSCs to form an interconnected, viable network that completely covers the ceramic surface [50].
- Step 2 - Hematopoietic Seeding: Primary human BM-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) are seeded into the scaffold, which is now populated with MSCs.
- Step 3 - On-Chip Dynamic Culture: The scaffold containing both MSCs and HSPCs is transferred into the microfluidic chip, and dynamic perfusion culture is initiated and maintained for up to 31 days. The perfusion of culture medium delivers nutrients and applies subtle fluidic forces [50].
- Step 4 - Differentiation and Analysis: The medium is supplemented with defined cytokine cocktails to direct multilineage hematopoietic differentiation. Non-adherent cells output from the scaffold are harvested from the medium compartment at regular intervals (e.g., days 10, 17, 24, 31) for analysis by flow cytometry and other functional assays [50].
A key advantage of this system is that the pre-cultured MSCs on the ceramic scaffold maintain a stable osteogenic phenotype under dynamic culture conditions, as confirmed by RNA sequencing showing enrichment for osteoblast signatures and pathways related to bone formation and extracellular matrix collagen deposition [50].
Functional Applications in Drug Safety and Disease Modeling
The physiological relevance of Bone Marrow-on-a-Chip systems makes them particularly valuable for the safety assessment of novel therapeutics, especially biologics, where traditional animal models often fail due to species-specific differences [48] [50].
Table 2: Modeling Hematopoietic Toxicity and Response on a Chip
| Therapeutic Class | Example Agent | Observed Effect in Bone Marrow-on-a-Chip | Clinical Relevance |
|---|---|---|---|
| Chemotherapy/Radiation | Clinically relevant doses | Lineage-specific depletion of blood cells (myelosuppression) [48] | Recapitulates dose-limiting toxicity in patients [48] |
| Transferrin Receptor-Targeting Antibody | IgG1 antibody | Inhibition of on-chip erythropoiesis [50] | Models mechanism-based anemia [50] |
| T Cell Bispecific Antibodies | Anti-CD3 Engager | T cell activation and target cell killing in an autologous setup [50] | Models on-target, off-tumor immunotoxicity [50] |
| Inherited Bone Marrow Failure Syndrome | Shwachman-Diamond Syndrome patient cells | Reproduced hallmark features, including impaired neutrophil maturation [48] | Provides a platform for studying patient-specific pathophysiology [48] |
These systems can also be rendered immunocompetent. By incorporating autologous peripheral blood T cells into the co-culture, researchers demonstrated the model's ability to recapitulate T cell-activating immunotherapy responses, including targeted killing and cytokine release, providing a tool for evaluating both efficacy and immune-related adverse events [50].
Direct Comparison: BMOs vs. Bone Marrow-on-a-Chip
The choice between using BMOs or a Bone Marrow-on-a-Chip depends on the specific research objectives, as each platform offers distinct advantages and has inherent limitations.
Table 3: Comparison of Bone Marrow Organoid and On-a-Chip Platforms
| Feature | Bone Marrow Organoids (BMOs) | Bone Marrow-on-a-Chip |
|---|---|---|
| Source Cells | Human induced pluripotent stem cells (iPSCs) [49] | Primary human cells (MSCs, CD34+ HSPCs) [50] |
| Self-Organization | High; spontaneous 3D assembly from iPSCs [49] | Engineered; dependent on scaffold design and cell seeding [50] |
| Key Microenvironmental Features | Endogenous generation of vascular networks, CAR cells, Nestin+ MSCs [49] | Perfusion, ceramic scaffold, engineered osteogenic niche [50] |
| Throughput | Moderate to High (96-well format) [49] | Lower throughput, but newer high-throughput chips are emerging [51] |
| Culture Duration | At least 60 days [49] | At least 31 days [50] |
| Primary Applications | Developmental studies, genetic disease modeling [49] | Drug safety profiling, toxicity testing, immune-oncology [48] [50] |
| Key Readouts | Single-cell RNA-seq, spatial mapping, engraftment potential [49] | Flow cytometry of output cells, cytokine measurement, functional immune assays [50] |
The Scientist's Toolkit: Essential Research Reagents
The successful implementation of these advanced models relies on a carefully selected toolkit of biological and engineering components.
Table 4: Essential Reagent Solutions for Bone Marrow Models
| Reagent / Material | Category | Critical Function in the Model | Specific Examples |
|---|---|---|---|
| Induced Pluripotent Stem Cells (iPSCs) | Starting Cell Source | Provides a genetically defined, patient-specific foundation for generating all cell types in BMOs [49] | Healthy donor or patient-specific iPSC lines [49] |
| Primary CD34+ HSPCs | Starting Cell Source | Provides authentic hematopoietic stem and progenitor cells for on-a-chip models [50] | Human bone marrow or mobilized peripheral blood-derived CD34+ cells [50] |
| Primary MSCs | Starting Cell Source | Forms the supportive stromal niche; secretes key maintenance factors [50] | Human bone marrow-derived mesenchymal stromal cells [50] |
| Defined Cytokine Cocktail | Soluble Factors | Directs differentiation and maintains/expands specific hematopoietic lineages [49] [50] | TPO, FLT-3L, SCF, EPO, IL-3, G-CSF, GM-CSF, IL-7, IL-15 [49] [50] |
| Microfluidic Chip & Scaffold | Engineering Material | Provides the 3D physical structure and enables perfusion culture [50] | Zirbonium oxide ceramic scaffold in HUMIMIC Chip2 [50]; High-throughput PDMS chips [51] |
| Extracellular Matrix (ECM) | Scaffolding Material | Supports 3D cell growth and self-organization; presents biochemical and mechanical signals [49] [47] | Collagen I, Matrigel [49] |
| Small Molecule Inducers | Signaling Modulators | Directs lineage specification during differentiation from iPSCs [49] | CHIR99021 (Wnt agonist), BMP4, SB431542 (TGF-β inhibitor) [49] |
Signaling Pathways Regulating the HSC Niche
The regulatory circuitry within the HSC niche is governed by a network of conserved signaling pathways. The following diagram synthesizes key ligand-receptor interactions between HSCs and niche cells, as identified in the research [46] [49] [47].
The experimental workflows for generating these two model systems are distinct, reflecting their different foundational principles. The following diagram outlines the parallel paths for creating BMOs from iPSCs and a Bone Marrow-on-a-Chip from primary cells.
Bone marrow organoids and Bone Marrow-on-a-Chip platforms represent a paradigm shift in hematopoietic research. BMOs, derived from iPSCs, offer a powerful, self-organizing model ideal for probing developmental biology and the mechanistic underpinnings of genetic blood disorders [49]. In contrast, microfluidic chips, often populated with primary cells, provide a controlled, perfusable system with demonstrated immediate utility in the functional safety profiling of novel therapeutics, including immunotherapies [48] [50]. Together, these complementary technologies provide a more physiologically relevant and human-specific toolkit than previously available, enabling deeper investigation into the complexities of the bone marrow microenvironment. Their continued development and integration hold the promise of accelerating drug discovery, personalizing treatment strategies, and fundamentally advancing our understanding of hematopoiesis in health and disease.
The hematopoietic stem cell (HSC) niche within the bone marrow represents a quintessential example of a complex, multi-faceted microenvironment that is indispensable for the regulation of stem cell fate. This dynamic three-dimensional (3D) unit provides a specialized anatomical site that not only supports the maintenance and survival of HSCs but also actively governs their self-renewal, differentiation, and quiescence through an intricate interplay of biochemical and biophysical signals [7] [52]. The physiological relevance of this niche extends beyond homeostasis, as its disruption plays a significant role in the etiology of various hematological disorders [7].
The clinical motivation for replicating this niche in vitro is compelling. HSC transplantation remains a cornerstone therapy for hematologic diseases, yet its efficacy is constrained by the scarcity of suitable HSC sources [7] [53]. In vitro expansion of HSCs from sources like umbilical cord blood (UCB) has proven challenging because HSCs rapidly lose their stemness in conventional two-dimensional (2D) culture systems, which fail to recapitulate the native niche [53] [54]. Furthermore, regulatory agencies are increasingly advocating for advanced alternative platforms that better emulate human physiology for drug safety evaluation, moving away from conventional animal testing [7]. Consequently, the biofabrication of 3D biomimetic models via tools like 3D bioprinting and advanced biomaterial scaffolds has emerged as a critical interdisciplinary endeavor, aiming to bridge the gap between in vivo complexity and in vitro controllability for applications in disease modeling, drug screening, and regenerative medicine [7] [55].
Deconstructing the Native HSC Niche: Key Components for Reconstruction
A systematic engineering of the HSC niche in vitro requires a thorough deconstruction of its native in vivo constituents. The bone marrow niche is not a singular entity but a coordinated assembly of sub-compartments, primarily the endosteal niche and the perivascular niche, which together form a network of extrinsic factors regulating HSC fate [53] [52] [54].
Core Cellular Components and Signaling Pathways
The functionality of the HSC niche is orchestrated by a diverse consortium of resident cells. Osteolineage cells were among the first identified niche components, contributing to the retention of multipotent progenitors (MPPs) and common lymphoid progenitors (CLPs) through secretion of factors like SDF-1 (CXCL12) [7] [52]. Endothelial cells forming the sinusoids are crucial for HSC maintenance and quiescence, notably as a specific cellular source of stem cell factor (SCF) [7]. Furthermore, non-hematopoietic cells such as mesenchymal stromal cells (MSCs) and CXCL12-abundant reticular (CAR) cells provide essential support through the secretion of SCF, CXCL12, and angiopoietin-1 (ANG-1) [53] [54].
Importantly, regulation also comes from hematopoietic-derived cells themselves. Megakaryocytes regulate HSC maintenance by secreting cytokines such as TPO, CXCL4, and TGF-β, and through physical interactions with osteomacs and osteoblasts [7]. Regulatory T cells (Tregs) contribute to establishing an immune-privileged site for HSCs, particularly in aged populations [7] [52]. The signaling output of a pathway is often determined by the cellular origin of the ligand; for instance, osteolineage-derived SDF-1 regulates progenitor retention, while endothelial-derived SDF-1 is critical for HSC quiescence [7]. This cellular complexity necessitates strategic selection of co-culture components in engineered niches.
Biophysical and Biochemical Properties of the Niche
The HSC niche is a physically defined space where mechanical properties are as instructive as chemical signals. The extracellular matrix (ECM) provides structural integrity and biochemical cues. It is predominantly composed of structural proteins (e.g., collagen I, III, IV, V, VI; fibronectin; laminin) and glycosaminoglycans (GAGs) such as hyaluronic acid (HA) and heparan sulfate proteoglycans (HSPGs) [7] [53]. HSCs perceive ECM cues through receptors like integrins, which mediate cell adhesion, migration, and differentiation [53].
A critical biophysical parameter is matrix stiffness, which is highly heterogeneous within the bone marrow. Measurements indicate that the endosteal niche is relatively rigid (>35 kPa), the vascular walls exhibit intermediate stiffness (5–8 kPa), and the central marrow is soft (~0.3 kPa) [7] [41]. This stiffness gradient mediates distinct HSC states; for example, softer substrates akin to the vascular niche have been shown to promote the maintenance of primitive HSC phenotypes, while stiffer environments often induce differentiation [54] [41]. Other vital physical parameters include oxygen tension (pO2), which forms a hypoxic gradient crucial for maintaining HSC quiescence, and fluid shear stress, which influences HSC behavior through dynamic mechanical forces from blood flow and interstitial fluid movement [7] [41].
Table 1: Key Biophysical Parameters of the Native HSC Niche and Their Functional Impact
| Parameter | In Vivo Characteristics | Impact on HSC Fate |
|---|---|---|
| Matrix Stiffness | Heterogeneous: Endosteal niche >35 kPa; vascular walls 5-8 kPa; central marrow ~0.3 kPa [7] [41] | Softer matrices support quiescence and stemness; stiffer matrices promote differentiation [54] [41]. |
| Oxygen Tension | Hypoxic gradient; higher pO2 (1.8%) near endosteum, lower (1.3%) in regions >40 μm away [53] | Hypoxia maintains HSC quiescence and preserves long-term repopulating capacity [41]. |
| ECM Topography | Nanofibrous architecture composed of collagen, fibronectin, laminin [53] [41] | Nanofibrous structures enhance cell-ECM interactions, promoting homing and retention. |
| Fluid Shear Stress | Induced by blood flow and marrow interstitial fluid movement [7] | Regulates HSC survival, proliferation, and lineage specification; activates mechanosensitive pathways [7]. |
Biomaterial Scaffolds for a 3D Microenvironment
The transition from 2D to 3D culture systems is a foundational step in HSC niche reconstruction. Three-dimensional biomaterial scaffolds provide the structural and mechanical framework that more accurately mimics the in vivo environment, thereby promoting more physiologically relevant HSC behavior.
Material Selection and Scaffold Design
The choice of biomaterial is paramount and often leans towards natural polymers or hybrid composites due to their inherent bioactivity. Key materials include:
- Hyaluronic Acid (HA): A major GAG in the bone marrow, HA is an excellent base for bioinks. Its derivatives can be functionalized to create hydrogel scaffolds that support HSC expansion and differentiation [7] [56].
- Fibrin: A natural hydrogel derived from fibrinogen, it is often used in conjunction with other materials to create supportive 3D environments for HSCs and megakaryocytes [41].
- Alginate: A seaweed-derived polymer valued for its biocompatibility and tunable physical properties via ionic crosslinking. It is widely used in 3D bioprinting and scaffold fabrication [41].
- Synthetic Polymers (e.g., PLA, PVA): These offer superior control over mechanical properties and degradation rates but typically lack innate cell adhesion motifs, requiring functionalization with ECM proteins or peptides like RGD [41].
The architectural design of the scaffold—including porosity, pore size, and interconnectivity—is critical for nutrient diffusion, waste removal, and cell migration. Optimal pore sizes for bone marrow-mimetic scaffolds typically range from 100 to 800 μm in diameter, which facilitates vascularization and efficient mass transport [41]. Furthermore, the surface topography of the scaffold, such as nanofibrous structures created by electrospinning, can mimic the native ECM and significantly influence HSC morphology and fate decisions [53] [41].
Functionalization with Biochemical Cues
To transform an inert scaffold into a bioactive niche, strategic functionalization with biochemical signals is required. This involves the incorporation of:
- ECM Proteins and Peptides: Coating or covalently conjugating scaffolds with fibronectin, laminin, or collagen provides specific binding sites for HSCs via integrin receptors. Short peptide sequences like RGD (Arg-Gly-Asp), derived from fibronectin, are commonly used to promote cell adhesion [53] [41].
- Cytokines and Growth Factors: The sustained presentation of key hematopoiesis-regulating factors is crucial. These include SCF, CXCL12, TPO, FLT3-L, and IL-6 [53] [54]. These molecules can be physically entrapped within the scaffold or chemically tethered to the material to create a stable, long-lasting gradient that guides HSC behavior.
- Co-culture with Niche Cells: Perhaps the most powerful method of functionalization is to populate the scaffold with supporting cells from the native niche, such as MSCs, osteoblasts, or endothelial cells [7] [41]. These cells create a living, dynamic microenvironment by secreting a complex cocktail of factors and directly interacting with HSCs, thereby more authentically recapitulating the in vivo niche.
Table 2: Summary of Key Biomaterials and Functionalization Strategies for HSC Niche Engineering
| Material/Strategy | Key Characteristics | Application in HSC Niche Engineering |
|---|---|---|
| Hyaluronic Acid (HA) | Natural polymer; major bone marrow GAG; tunable mechanical properties [56]. | Base for bioinks; supports 3D cell encapsulation and printing of stromal cells [56]. |
| Fibrin | Natural hydrogel; excellent biocompatibility; contains cell adhesion sites. | Used in 3D cultures for megakaryocyte function and platelet production [41]. |
| Alginate | Natural polymer; ionically crosslinkable; highly tunable stiffness. | Used in 3D bioprinting and for creating scaffolds to study HSC differentiation. |
| Synthetic Polymers (PLA, PVA) | Precisely controllable mechanics and degradation; reproducible. | Provide structural support in composite scaffolds; often requires RGD functionalization [41]. |
| ECM Functionalization | Coating with fibronectin, laminin, collagen, or RGD peptides. | Enhances HSC adhesion, homing, and can direct lineage-specific differentiation [53] [41]. |
| Cytokine Delivery | Sustained release of SCF, TPO, CXCL12, FLT3-L. | Promotes HSC maintenance, self-renewal, and expansion in 3D culture [53] [54]. |
| Stromal Co-culture | Co-culture with MSCs, osteoblasts, endothelial cells. | Creates a dynamic, self-renewing signaling environment that best maintains HSC stemness ex vivo [7] [41]. |
3D Bioprinting the Bone Marrow Niche
3D bioprinting represents the vanguard of HSC niche reconstruction, offering unprecedented spatial control over the placement of cells and biomaterials to create complex, biomimetic 3D architectures.
Bioprinting Technologies and Workflows
The most common bioprinting modality for soft tissues like bone marrow is extrusion-based printing. This method utilizes a pneumatic or mechanical (piston/screw) dispensing system to continuously extrude a filament of a bioink—a printable combination of biomaterials and living cells [56] [57]. The process for creating a bioprinted bone marrow model typically follows a structured workflow, as illustrated below.
Advanced Bioink Engineering
The bioink is the cornerstone of successful bioprinting. An ideal bioink must be printable, provide mechanical support, and be cytocompatible. Recent advances have led to the development of sophisticated, niche-specific bioinks. A prime example is the dual-functionalized hyaluronic acid (HA) bioink. This involves a "one-pot synthesis" where HA is modified with both methacrylate groups (for covalent photochemical cross-linking to ensure structural stability) and alkyl side chains (for physical cross-linking via hydrophobic interactions, providing self-healing and shear-thinning properties) [56]. This combination yields a bioink that flows under the shear stress of printing but rapidly recovers its shape afterwards, enabling the printing of complex structures without additional viscosity enhancers that could compromise cell viability [56].
Post-printing, two primary strategies for cell incorporation are used: cell encapsulation (mixing cells homogeneously within the bioink prior to printing) and cell injection (seeding cells into pre-printed and stabilized channels or chambers post-fabrication) [56]. The latter allows for the precise placement of different cell types, such as HSCs and stromal cells, into distinct but adjacent locations within the construct, mirroring the spatial organization of the native niche.
Experimental Protocols for Niche Evaluation
Robust and standardized protocols are essential for the fabrication and functional validation of engineered HSC niches. Below is a detailed methodology for a representative experiment: creating a 3D bioprinted bone marrow model for drug testing.
Protocol: 3D Bioprinting of a Miniaturized Bone Marrow Niche for High-Throughput Drug Screening
Objective: To fabricate a vascularized human bone marrow organoid containing HSPCs and stromal cells for in vitro modeling of normal and malignant hematopoiesis and subsequent drug efficacy/toxicity testing.
Materials and Reagents:
- Cells: CD34+ HSPCs (from cord blood or mobilized peripheral blood), Human Bone Marrow-derived Mesenchymal Stromal Cells (BM-MSCs), Human Umbilical Vein Endothelial Cells (HUVECs).
- Bioink: Dual-functionalized Hyaluronic Acid (HA) bioink [56].
- Culture Media: Serum-free HSPC expansion medium, MSC growth medium, Endothelial Cell Growth Medium.
- Bioprinter: Extrusion-based 3D bioprinter (e.g., custom-built or commercial equivalent) equipped with a 22G-27G printhead [56] [57].
- Other Reagents: Photoinitiator (e.g., LAP), UV light source (365 nm, 5-10 mW/cm²) for crosslinking, sterile PBS.
Procedure:
- Pre-bioprinting Processing:
- Cell Expansion: Expand and passage HUVECs and BM-MSCs in their respective media. Expand CD34+ HSPCs in cytokine-enriched serum-free medium (SCF, TPO, FLT3-L) for 3-5 days.
- Bioink Preparation: Thaw or prepare the dual-functionalized HA bioink. Sterilize if necessary. Mix the bioink with a photoinitiator at a cytocompatible concentration (e.g., 0.1% w/v LAP).
- Stromal Cell-Laden Bioink: Mix a portion of the prepared bioink with BM-MSCs and HUVECs to a final density of 10-20 x 10^6 cells/mL. Keep on ice to prevent premature gelation.
Bioprinting Process:
- Load the cell-laden bioink into a sterile printing cartridge.
- Set the bioprinter parameters: Pressure 15-30 kPa, printing speed 5-10 mm/s, nozzle size 22G-27G. Print at a temperature of 4-18°C to maintain cell viability and optimize ink viscosity.
- Print the desired structure (e.g., a grid or honeycomb lattice) layer-by-layer onto a heated (37°C) print bed.
- After each layer, apply a brief, low-intensity UV light exposure (e.g., 30-60 seconds) for partial crosslinking to ensure layer adhesion and structural fidelity.
Post-Printing Processing and Culture:
- After the final layer is printed, perform a final full crosslinking step with UV light for 2-5 minutes.
- Transfer the printed constructs to culture plates and wash with PBS to remove any residual photoinitiator.
- HSPC Seeding: Seed CD34+ HSPCs (1-5 x 10^5 cells/construct) onto the surface of the printed scaffold or inject them into pre-designed channels within the structure [56].
- Maintain the constructs in a mixed medium (e.g., 1:1 ratio of HSPC and MSC media) in a humidified incubator at 37°C and 5% CO2. Change the medium every 2-3 days.
Functional Assays and Readouts:
- Long-term Culture-Initiating Cell (LTC-IC) Assay: After 3-6 weeks of culture, dissociate the constructs and plate cells in methylcellulose to quantify primitive progenitor cells [7].
- Flow Cytometry: Analyze cell populations for markers of HSC stemness (e.g., CD34+CD38-, CD90+), differentiation (lineage markers), and viability at multiple time points.
- Confocal Microscopy: Image fixed and stained constructs to visualize 3D spatial organization of HSCs relative to stromal cells and vascular networks using antibodies against CD34, CD31, and other niche markers [58].
- Drug Testing: Expose the mature constructs to chemotherapeutic agents or novel compounds. Monitor HSPC viability, proliferation, and differentiation compared to untreated controls, effectively creating a "clinical trial in a dish" [57].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table catalogs key reagents and materials essential for the biofabrication and analysis of engineered HSC niches, as derived from the cited research.
Table 3: Research Reagent Solutions for HSC Niche Biofabrication
| Item | Function/Application | Specific Examples & Notes |
|---|---|---|
| Dual-functionalized HA Bioink | Base material for 3D bioprinting bone marrow constructs. Provides a biomimetic, printable hydrogel. | HA modified with methacrylate (covalent crosslinking) and alkyl chains (physical crosslinking). Allows printing without additives [56]. |
| CD34+ HSPCs | Target functional cell population for expansion and study within the niche. | Sourced from umbilical cord blood, mobilized peripheral blood, or bone marrow. Identified by CD34+CD38- phenotype [7] [53]. |
| Stromal Co-culture Cells | Provide essential physiological cues to support HSCs. | Mesenchymal Stromal Cells (MSCs), Endothelial Cells (HUVECs), Osteoblasts (OBs). Can be pre-seeded in scaffolds or printed simultaneously [7] [41]. |
| Key Recombinant Cytokines | Soluble biochemical signals to maintain HSC stemness and promote survival. | Stem Cell Factor (SCF), Thrombopoietin (TPO), Fms-like tyrosine kinase 3 ligand (FLT3-L), CXCL12 (SDF-1) [53] [54]. |
| ECM Protein Coatings | Functionalize scaffold surfaces to enhance cell adhesion and mimic native ECM. | Fibronectin, Laminin, Collagen I/IV. Coating stiffness and composition can direct lineage specification [53] [41]. |
| Photoinitiator (LAP) | Enables covalent crosslinking of methacrylated bioinks (e.g., GelMA, HAMA) via UV light. | Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP). Offers superior cytocompatibility compared to older initiators [56]. |
The field of HSC niche biofabrication is progressing at an accelerated pace, driven by the convergence of advanced biomaterials, precision bioprinting, and deep biological insights. The development of sophisticated in vitro models, from functionalized 3D scaffolds to bioprinted living organoids, is steadily closing the fidelity gap with the native bone marrow microenvironment [7] [55]. These engineered systems are already enabling unprecedented studies in normal and malignant hematopoiesis, high-throughput drug screening, and the ex vivo expansion of HSCs for therapeutic applications [7] [58].
However, significant challenges remain. The heterogeneity and dynamic nature of the in vivo niche are difficult to fully capture. Future efforts will need to focus on integrating more complex multicellular communities, including immune and neural cells, and creating even more physiologically accurate vascular networks [7] [57]. Furthermore, the standardization of culture protocols and the seamless integration of emerging technologies like artificial intelligence (AI) for design optimization and multi-omics analyses for rigorous model validation will be critical for the widespread adoption and clinical translation of these powerful biofabrication tools [7]. As these technologies mature, they hold the definitive promise to revolutionize both our fundamental understanding of hematopoiesis and the clinical management of blood disorders.
The hematopoietic stem cell (HSC) niche is a highly complex and dynamically regulated microenvironment that supports the survival, self-renewal, and differentiation of blood stem cells. Recent technological advances have enabled the reconstruction of this niche in vitro, moving from conventional two-dimensional (2D) cultures toward sophisticated three-dimensional (3D) biomimetic models. These innovations have unlocked critical applications in HSC expansion, lineage-specific differentiation, and high-throughput drug discovery, providing powerful platforms for both basic research and clinical translation. This technical guide explores the current methodologies, experimental protocols, and research tools that are reshaping this rapidly evolving field, framed within the broader context of HSC niche and bone marrow microenvironment research.
Platform Technologies for HSC Niche Reconstruction
The fidelity of in vitro HSC models to their native in vivo counterparts has been progressively enhanced through the integration of cutting-edge bioengineering platforms [28] [59]. The table below summarizes the primary technologies currently employed:
Table 1: Platform Technologies for In Vitro HSC Niche Reconstruction
| Platform Type | Key Characteristics | Primary Applications | Technical Considerations |
|---|---|---|---|
| 2D Co-culture Systems | Conventional monolayer cultures with stromal feeder cells; technically simple | Preliminary HSC maintenance studies; basic differentiation assays | Limited physiological relevance; lacks 3D architecture |
| 3D Biomimetic Models | Hydrogel-based (e.g., GelMA, Gel-HA) scaffolds mimicking ECM; spheroid cultures | Enhanced HSC expansion; study of cell-ECM interactions | Optimization of scaffold composition and stiffness required |
| Bone Marrow Organoids (BMOs) | Self-organizing 3D structures containing multiple niche cell types | Disease modeling; study of niche cell crosstalk; developmental biology | Challenges in standardization and reproducibility |
| Bone Marrow-on-a-Chip | Microfluidic devices with perfusable vascular channels; dynamic culture conditions | Drug toxicity testing; hematopoiesis studies under flow conditions | Requires specialized equipment and expertise |
| 3D Bioprinted Niches | Precise spatial patterning of niche cells and matrix components using bioinks | Customizable niche architectures; high-throughput screening | Resolution limitations; viability maintenance during printing |
The shift from 2D to 3D culture systems represents a paradigm shift in HSC research, with 3D biomimetic models demonstrating superior capacity for maintaining stemness and enabling multilineage differentiation through better replication of native biomechanical and biochemical cues [28].
Experimental Protocols for Key Applications
HSC Expansion from Induced Pluripotent Stem Cells (iPSCs)
A groundbreaking protocol for generating functional HSCs from human iPSCs was recently published, achieving robust long-term multilineage engraftment in immunodeficient mice [60]. The step-by-step methodology is outlined below:
Table 2: Step-by-Step Protocol for iPSC to iHSC Differentiation
| Stage | Duration | Key Media Components | Growth Factors/Cytokines | Purpose |
|---|---|---|---|---|
| Mesoderm Induction | 24 hours | Defined base medium | 4 µM CHIR99201 (Wnt agonist), 3 ng/ml BMP4, 5-30 ng/ml Activin A | Specifies mesodermal lineage |
| HOXA Patterning | 2 days | Defined base medium | CHIR99201, BMP4, Activin A | Patterns mesoderm to AGM-like, HOXA+ state |
| Hemogenic Endothelium Specification | 4 days (days 3-7) | Defined base medium | BMP4, VEGF; Retinyl acetate (RETA) | Specifies hemogenic endothelium; critical for MLE capacity |
| Endothelial-to-Hematopoietic Transition | 7 days (days 7-14) | Defined base medium | VEGF removal | Initiates blood cell emergence; mimics intra-arterial clusters |
| Cell Harvest & Cryopreservation | Day 14-16 | Cryopreservation medium | - | Harvest of CD34+ cells for transplantation or testing |
This protocol successfully generated functionally defined, multipotent CD34+ hematopoietic cells (iHSCs) that produced engraftment levels similar to umbilical cord blood transplantation, with 25-50% of immune-deficient recipient mice showing multilineage engraftment [60]. The timed provision of Wnt agonists, retinoic acid precursors (retinyl acetate), and VEGF proved critical for endowment with repopulating capacity.
Diagram 1: iPSC to iHSC Differentiation Workflow
High-Throughput Screening for Hematopoietic Differentiation
Traditional clonogenic "colony assays" for assessing hematopoietic progenitor differentiation have extremely low throughput, precluding their use in library screening and extensive drug discovery work [61]. A rapid-throughput alternative methodology has been developed:
CELISA (Cell-Based Enzyme-Linked Immunosorbent Assay) Protocol:
- Cell Culture Setup: Plate hematopoietic progenitor cells in 96-well filter plates compatible with cell culture.
- Compound Screening: Treat with drug candidates or experimental conditions according to screening library design.
- Lineage Marker Quantification: Incubate with lanthanide-conjugated primary antibodies against hematopoietic lineage-specific markers (myeloid, erythroid, megakaryocytic).
- Signal Detection: Perform time-resolved fluorescence spectroscopy on the intact cells.
- Data Analysis: Quantify differentiation outcomes based on lineage marker expression profiles.
This assay requires approximately 1 hour for quantitation and demonstrates excellent correlation with data generated using the traditional colony assay, making it suitable for both rapid-throughput drug discovery and toxicity screening in hematopoiesis research [61].
Neutrophil Differentiation for Disease Modeling
A comprehensive protocol for human neutrophil differentiation from CD34+ hematopoietic stem and progenitor cells (HSPCs) provides a model for studying granulopoiesis and neutrophil function across physiological and pathological contexts [62]:
- CD34+ HSPC Isolation: Isolate cells from bone marrow through Ficoll gradient centrifugation followed by magnetic bead-based separation using the EasySep Human CD34+ Cell Selection Kit II.
- Expansion Culture: Maintain cells in StemSpan SFEM II hematopoietic stem cell medium supplemented with IL-3, IL-6, TPO, SCF, and FLT-3L.
- Myeloid Expansion Phase (Days 0-7): Culture in RPMI 1640 with 10% FCS, SCF, IL-3, and G-CSF, changing medium every two days.
- Granulocytic Differentiation (Day 8+): Switch to granulocytic differentiation medium (RPMI 1640 with 10% FCS and G-CSF).
- Functional Patterning (Day 13+): Stimulate with specific cytokines (TGF-β, IFNβ, or GM-CSF) to drive toward distinct functional states (pro-inflammatory, cancer-associated, or mature neutrophil states).
This protocol enabled the identification of IFNB, GMCSF, and TGFB as drivers of distinct neutrophil states and revealed the transcription factor JUNB as a driver of angiogenic and immunosuppressive neutrophil functions [62].
Signaling Pathways in HSC Niche Regulation
The HSC niche is regulated by complex signaling interactions that can be targeted for in vitro manipulation. Recent research has identified key pathways with therapeutic potential:
Diagram 2: Rhosin-Mediated HSC Rejuvenation Pathway
RhoA Pathway Inhibition for HSC Rejuvenation: Research has identified that RhoA, a mechanosensor protein, becomes highly activated as blood stem cells age, leading to nuclear envelope stress and epigenetic dysfunction [63]. The small molecule Rhosin (a RhoA inhibitor) has demonstrated capacity to reverse age-associated changes in HSCs through:
- Reduction of mechanostress on the nuclear envelope
- Improvement of chromatin organization and epigenetic marker preservation
- Enhanced regenerative capacity of the immune system
- Improved blood cell production following transplantation
This rejuvenation strategy targets the core ageing process rather than merely combating its effects, representing a paradigm shift in approaches to age-related haematopoietic decline [63].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for HSC Niche Studies
| Reagent Category | Specific Products | Research Applications | Technical Notes |
|---|---|---|---|
| HSC Sources | Mobilized peripheral blood, bone marrow, umbilical cord blood, iPSCs | Provide primary HSCs for research; iPSCs enable genetic manipulation | CD34+ selection common for human HSCs [64] |
| Culture Media | StemPro-34 SFM, StemSpan SFEM II | Serum-free HSC expansion; iPSC to HSC differentiation | Supports primitive hematopoietic populations [64] [60] |
| Cytokines & Growth Factors | SCF, FLT3-L, TPO, IL-3, IL-6, G-CSF, BMP4, VEGF | Lineage-specific differentiation; HSC self-renewal; niche patterning | Retinoids (retinyl acetate) critical for engraftment capacity [60] |
| Gene Editing Tools | Lipid-based transfection, electroporation, viral delivery systems (lentiviral, retroviral) | Genetic manipulation; disease modeling; gene correction | CRISPR/Cas9 compatible systems |
| Lineage Markers | CD34, CD38, CD45, CD90, CD133, lineage-specific antigens | HSC identification; differentiation validation; purity assessment | CD34+ common for human HSC identification [64] |
| Small Molecule Modulators | CHIR99201 (Wnt agonist), SB431542 (ALK inhibitor), Rhosin (RhoA inhibitor) | Pathway manipulation; HSC expansion; rejuvenation studies | Rhosin shows rejuvenation potential [63] |
Challenges and Future Directions
Despite significant advances, several challenges persist in the field of in vitro HSC niche reconstruction. Standardization of culture protocols across laboratories remains difficult, impacting reproducibility and comparability of results [28]. The integration of emerging technologies such as single-cell multi-omics, advanced microscopy, and artificial intelligence presents both opportunities and implementation challenges [28] [59]. Most importantly, the translation of these innovations into clinical practice requires rigorous validation and scaling of production processes to meet therapeutic requirements [28].
Future research directions will likely focus on enhancing the physiological relevance of in vitro models through incorporation of additional niche components, including immune cells, neural inputs, and mechanical signaling cues. The application of machine learning for analyzing complex multidimensional data from these systems will accelerate the identification of key regulatory mechanisms and potential therapeutic targets [59] [63]. As these technologies mature, they promise to revolutionize both our understanding of hematopoiesis and our capacity to treat blood disorders through regenerative medicine approaches.
The hematopoietic stem cell (HSC) niche within the bone marrow represents a highly complex and dynamically regulated microenvironment that controls stem cell fate decisions, including self-renewal, quiescence, and differentiation. Recent technological advancements have enabled unprecedented insights into this intricate system. This whitepaper provides an in-depth technical guide to the integrative methodologies—multi-omics, advanced microscopy, and artificial intelligence—that are revolutionizing the validation of HSC niche models. We detail specific experimental protocols, present quantitative data comparisons, and visualize core workflows and signaling pathways. For researchers and drug development professionals, this resource offers a comprehensive toolkit for implementing these cutting-edge technologies in hematopoietic microenvironment research.
The hematopoietic stem cell (HSC) niche is a specialized bone marrow microenvironment that regulates the maintenance, self-renewal, and differentiation of HSCs through complex biochemical and biophysical signals [65] [7]. This niche encompasses various cellular components, including osteoblasts, endothelial cells, mesenchymal stem cells (MSCs), and perivascular stromal cells (PerSCs), embedded within a specialized extracellular matrix (ECM) [7] [39]. A critical challenge in HSC research has been accurately replicating this native microenvironment in vitro to enable reliable disease modeling and drug screening.
Traditional two-dimensional (2D) culture systems fail to recapitulate the three-dimensional (3D) architecture and physiological complexity of the bone marrow, leading to rapid HSC differentiation and loss of long-term reconstituting capacity [7] [39]. The validation of engineered HSC niches now requires sophisticated technologies that can capture the dynamic, multi-scale interactions within this system. The integration of multi-omics approaches, advanced microscopy, and artificial intelligence (AI) has emerged as a powerful paradigm for addressing these challenges, enabling comprehensive model validation across molecular, cellular, and tissue levels.
Multi-Omics Approaches for HSC Niche Deconstruction
Single-Cell Genomics and Transcriptomics
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity during HSC development and within the bone marrow niche. This technology enables the identification of rare cell populations, such as hemogenic endothelial cells (HECs) and pre-HSCs, which are critical for definitive hematopoiesis [13].
Protocol: Single-Cell RNA Sequencing of Bone Marrow Niches
- Tissue Dissociation: Obtain bone marrow biopsies and dissociate into single-cell suspensions using collagenase type IV (1-2 mg/mL) and DNase I (0.1 mg/mL) in PBS for 30-45 minutes at 37°C with gentle agitation.
- Cell Viability and Concentration: Filter cells through a 40-μm strainer and assess viability (>90% required) using trypan blue or automated cell counters.
- Cell Sorting: Enrich target populations (e.g., CD34+ HSCs, CD271+ mesenchymal stromal cells) using fluorescence-activated cell sorting (FACS) with antibodies against CD34-APC/Cy7 (1:100), CD45-BV510 (1:200), and CD271-PE (1:100).
- Library Preparation: Utilize droplet-based platforms (10x Genomics) according to manufacturer's protocols. Target 5,000-10,000 cells per sample with 50,000 reads per cell.
- Sequencing: Perform on Illumina platforms (NovaSeq 6000) with recommended depth of 50,000 reads per cell.
- Bioinformatic Analysis: Process data using Cell Ranger pipeline followed by Seurat or Scanpy for clustering, differential expression, and trajectory inference [13].
Table 1: Key Single-Cell Omics Applications in HSC Niche Research
| Application | Technology | Key Insights | References |
|---|---|---|---|
| Cellular Heterogeneity | scRNA-seq | Identified transition states from hemogenic endothelium to HSCs | [13] |
| Epigenetic Regulation | scATAC-seq | Mapped chromatin accessibility in HSC subpopulations | [66] |
| Spatial Organization | Spatial Transcriptomics | Resolved topographic distribution of niche cells | [67] |
| Clonal Evolution | scDNA-seq | Tracked clonal hematopoiesis in aging | [65] |
Integration of Multi-Omics Data
Systems biology (SysBio) approaches integrate multi-omics datasets to construct comprehensive regulatory networks governing HSC fate. Integrative analysis of transcriptomic, epigenomic, and proteomic data has revealed key transcription factors (GATA2, RUNX1, GFI1/GFI1B) and signaling pathways (Notch, Wnt/β-catenin, BMP) that orchestrate the endothelial-to-hematopoietic transition (EHT) during HSC emergence [13] [66].
Advanced Microscopy for Spatial and Dynamic Analysis
Super-Resolution and Multiplexed Imaging
Advanced microscopy techniques enable visualization of HSC-niche interactions at nanometer resolution and with multiple molecular targets. FLASH-PAINT, a recently developed super-resolution technique, uses transiently binding DNA imagers that allow virtually unlimited multiplexing capacity for visualizing complex subcellular structures [68].
Protocol: FLASH-PAINT for HSC Niche Imaging
- Sample Preparation: Fix bone marrow sections (4-8 μm) or whole-mount preparations with 4% PFA for 15 minutes at room temperature.
- Antibody Conjugation: Conjugate primary antibodies with docking DNA strands (5'-amine modification) using NHS ester chemistry. Recommended antibodies: anti-CD34 (HSC marker), anti-CD144 (endothelial cell marker), anti-Nestin (MSC marker), anti-Collagen IV (ECM marker).
- Imaging Probe Design: Design complementary DNA imagers (8-12 bp) conjugated with fluorescent dyes (Alexa Fluor 488, 568, 647). Use low concentrations (0.1-1 nM) to ensure transient binding.
- Sequential Imaging: Acquire images using a STORM/PAINT super-resolution microscope with 640 nm, 560 nm, and 488 nm laser lines. Apply bleaching steps between imaging cycles to remove residual signal.
- Image Reconstruction: Process raw data using open-source software (ImageJ, Picasso) to localize single molecules and reconstruct super-resolution images [68] [67].
Table 2: Advanced Microscopy Techniques for HSC Niche Analysis
| Technique | Resolution | Multiplexing Capacity | Applications in HSC Research | |
|---|---|---|---|---|
| Confocal Microscopy | ~250 nm | 4-5 channels | 3D reconstruction of bone marrow architecture | [67] |
| Two-Photon Microscopy | ~300 nm | 3-4 channels | Intravital imaging of HSC dynamics in living mice | [67] |
| STED Microscopy | ~30 nm | 2-3 channels | Nanoscale organization of membrane receptors | [69] |
| FLASH-PAINT | ~20 nm | Virtually unlimited | Mapping molecular interactions in niche cells | [68] |
| Holotomographic Microscopy | N/A (label-free) | N/A | 3D reconstruction of lipid bodies and organelles | [69] |
Intravital and 3D Imaging
Intravital microscopy through surgically implanted bone windows enables real-time observation of HSC behavior in living animals. This approach has revealed dynamic processes such as HSC mobilization, homing, and interactions with niche components under physiological conditions [67].
Artificial Intelligence and Machine Learning Applications
Predictive Modeling for HSC Behavior
Machine learning (ML) algorithms leverage high-dimensional data to predict HSC expansion, differentiation potential, and clinical outcomes. These models integrate donor characteristics, molecular profiles, and culture parameters to optimize HSC manipulation protocols [70] [66].
Protocol: Developing ML Models for HSC Mobilization Prediction
- Data Collection: Compile retrospective dataset including donor demographics (age, sex, BMI), baseline laboratory values (complete blood count, CD34+ count), and mobilization parameters (G-CSF dose, timing).
- Feature Engineering: Select relevant features using correlation analysis and domain knowledge. Normalize continuous variables and encode categorical variables.
- Model Training: Implement multiple algorithms (random forest, gradient boosting, support vector machines) using scikit-learn or TensorFlow. Use 5-fold cross-validation to assess performance.
- Model Validation: Evaluate models on held-out test sets using accuracy, precision, recall, and area under the ROC curve. The best-performing model in recent studies achieved 83% accuracy in predicting CD34+ cell yield after G-CSF mobilization [70].
- Clinical Implementation: Deploy the validated model as a web-based tool or integrated into electronic health record systems for donor screening and mobilization strategy selection.
AI-Enhanced Image and Data Analysis
Deep learning approaches, particularly convolutional neural networks (CNNs), automate the analysis of complex microscopy data, enabling high-throughput quantification of HSC-niche interactions. These algorithms can segment individual cells, classify morphological features, and track dynamic behaviors in time-lapse imaging [67] [66].
Table 3: AI/ML Applications in HSC Research and Clinical Translation
| Application | Algorithm Type | Function | Performance Metrics | |
|---|---|---|---|---|
| HSC Mobilization Prediction | Random Forest | Predicts CD34+ yield from donor characteristics | 83% accuracy, AUC 0.89 | [70] |
| Cell Classification | Convolutional Neural Network | Identifies HSC subpopulations from morphology | >90% precision | [66] |
| Drug Screening | Graph Neural Networks | Predicts compound effects on HSC expansion | R² = 0.79 for dose-response | [66] |
| Clinical Outcome Prediction | Survival Models | Estimates transplant success from multi-omics data | C-index = 0.75 | [66] |
Integrated Experimental Workflows for Model Validation
Validating Bioengineered HSC Niches
The integration of these technologies enables comprehensive validation of engineered HSC niches. For example, bioengineered niches that recreate physiological extracellular matrix organization can be assessed through multi-omics analysis of HSC transcriptional states, super-resolution microscopy of cell-ECM interactions, and AI-based tracking of long-term HSC maintenance [7] [39].
Protocol: Validating 3D Bioengineered HSC Niches
- Niche Fabrication: Create soft collagen type-I hydrogels (Young's modulus 1-10 kPa) with controlled fibrillar organization on poly(ethyl acrylate) surfaces to promote nestin expression in perivascular stromal cells.
- Functional Assessment: Seed CD34+ HSCs onto engineered niches and monitor:
- LT-HSC maintenance using long-term culture initiating cell (LTC-IC) assays
- Metabolic status through Seahorse extracellular flux analysis
- In vivo reconstitution capacity via transplantation into immunodeficient mice (NSG)
- Multi-Omics Validation: Perform scRNA-seq on retrieved cells to confirm preservation of LT-HSC transcriptional signature (e.g., upregulation of HLF, MLLT3, SPINK2).
- Structural Validation: Use FLASH-PAINT microscopy to visualize spatial relationships between HSCs and nestin+ stromal cells at 20-30 nm resolution.
- AI-Integrated Analysis: Apply machine learning models to integrate functional, molecular, and spatial data to predict niche efficacy and identify key quality attributes [39].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 4: Key Research Reagent Solutions for HSC Niche Studies
| Reagent/Platform | Type | Function | Example Applications | |
|---|---|---|---|---|
| Collagen Type-I Hydrogels | Biomaterial | Creates soft 3D microenvironment mimicking bone marrow stiffness | Maintaining LT-HSCs through nestin induction in PerSCs | [39] |
| Poly(ethyl acrylate) (PEA) | Polymer Surface | Promotes fibronectin unfolding and presentation of growth factors | Recapitulating endosteal surface for HSC maintenance | [39] |
| DNA Imagers (FLASH-PAINT) | Imaging Probe | Enables multiplexed super-resolution microscopy through transient binding | Visualizing multiple niche components simultaneously | [68] |
| CD34 MicroBead Kit | Cell Separation | Immunomagnetic selection of hematopoietic stem/progenitor cells | Isolation of HSCs from bone marrow or cord blood | [7] |
| StemRegenin-1 (SR1) | Small Molecule | Inhibits aryl hydrocarbon receptor to promote HSC expansion | Ex vivo expansion of HSCs in culture systems | [39] |
| Plerixafor (AMD3100) | CXCR4 Antagonist | Blocks SDF-1/CXCR4 axis to mobilize HSCs from niche | HSC mobilization for collection and transplantation | [70] |
The integration of multi-omics technologies, advanced microscopy, and artificial intelligence represents a paradigm shift in HSC niche research and model validation. These complementary approaches enable researchers to deconstruct the complexity of the bone marrow microenvironment across multiple scales—from molecular interactions to tissue-level organization. As these technologies continue to evolve, we anticipate increased standardization of validation protocols and more sophisticated computational models that can predict HSC behavior with greater accuracy.
The ongoing development of these integrative methodologies promises to accelerate the translation of engineered HSC niches into clinical applications, including expanded options for stem cell transplantation, improved disease modeling, and more effective drug screening platforms. For research and drug development professionals, mastering these technologies and their integrated implementation will be essential for advancing the next generation of hematopoietic stem cell therapies.
The Niche in Crisis: Inflammation, Remodeling, and Therapeutic Targeting in Blood Disorders
Inflammatory Stromal Cells (iMSCs) and the Feed-Forward Loop in MDS and CHIP
The bone marrow microenvironment (BMME) is no longer considered a passive bystander in hematopoietic pathogenesis but an active contributor to malignant evolution. Recent research has identified a distinct population of inflammatory mesenchymal stromal cells (iMSCs) that emerge in clonal hematopoiesis of indeterminate potential (CHIP) and expand in myelodysplastic syndromes (MDS). These iMSCs engage in a self-reinforcing inflammatory loop with immune cells, particularly interferon-responsive T cells, fundamentally remodeling the stem cell niche. This feed-forward circuit suppresses normal hematopoiesis, promotes vascular remodeling, and creates a permissive environment for clonal expansion, positioning the BMME as a critical therapeutic target for intercepting pre-malignant progression.
The bone marrow (BM) niche constitutes a specialized microenvironment where hematopoietic stem cells (HSCs) reside, self-renew, and differentiate. This complex cellular metropolis includes mesenchymal stromal cells (MSCs), endothelial cells, osteolineage cells, adipocytes, and immune cells that collectively regulate hematopoiesis through direct contact and secreted factors [71] [18]. With aging, the BM niche undergoes significant remodeling characterized by increased adipogenesis and chronic inflammation, creating conditions conducive to the emergence of CHIP [72] [20].
CHIP, characterized by somatic mutations in hematopoietic stem/progenitor cells (HSPCs) with variant allele frequency ≥2% in the absence of cytopenias or dysplasia, affects >10% of adults over 65 and confers increased risk for hematologic malignancies and cardiovascular disease [72] [71]. Myelodysplastic syndromes (MDS) represent a progression from this pre-malignant state, featuring ineffective hematopoiesis, cytopenias, and heightened risk of transformation to acute myeloid leukemia (AML) [73]. While driver mutations in HSPCs are necessary for CHIP and MDS pathogenesis, emerging evidence indicates that stromal inflammation actively shapes disease trajectory from its earliest stages [72] [19] [74].
Inflammatory Remodeling of the Bone Marrow Niche
The Emergence of Inflammatory Mesenchymal Stromal Cells (iMSCs)
Single-cell RNA sequencing of human bone marrow from a balanced cohort of 84 donors (35 age-matched controls, 17 CHIP carriers, and 32 MDS patients) revealed profound stromal remodeling across disease states [72]. This investigation identified a distinct population of inflammatory mesenchymal stromal cells (iMSCs) that arise in CHIP and become more prevalent in MDS, coinciding with the loss of CXCL12+ adipogenic stromal cells that normally support HSPC maintenance [72] [19].
Table 1: Stromal Cell Population Changes Across Disease States
| Cell Population | Healthy BM | CHIP | MDS | Functional Consequences |
|---|---|---|---|---|
| CXCL12⁺ Stromal Cells | Normal | Decreased | Significantly decreased | Loss of HSPC maintenance signals |
| Inflammatory MSCs (iMSCs) | Rare/absent | Emerge | Expand significantly | Create pro-inflammatory niche |
| Adipogenic Stromal Cells | Normal | Increased adipogenic shift | Further increased | Contribute to "fatty marrow" and inflammation |
Unlike their homeostatic counterparts, iMSCs exhibit a profound inflammatory signature, releasing large amounts of interferon-induced cytokines and chemokines that attract and activate immune cells [19] [74]. Functional studies in primary BM HSPC-MSC co-cultures demonstrated that healthy aged and CHIP HSPCs activate stromal support, while MDS HSPCs fail to do so [72]. Specifically, MDS HSPCs cannot induce stromal cells to produce CXCL12, a critical chemokine for hematopoietic homing and maintenance, potentially explaining the functional collapse of bone marrow in advanced disease [74] [75].
T Cell-Mediated Amplification and Feed-Forward Inflammation
The inflammatory reprogramming of the stromal compartment coincides with the expansion of IFN-responsive T cells that preferentially interact with iMSCs [72]. This creates a self-reinforcing circuit where iMSCs produce chemokines that recruit and activate T cells, which in turn release interferon-gamma and other inflammatory mediators that further stimulate iMSCs [19] [74].
Table 2: Inflammatory Mediators in the Remodeled Niche
| Inflammatory Component | Source | Key Effectors | Functional Role in Feed-Forward Loop |
|---|---|---|---|
| iMSC Secretome | Inflammatory MSCs | IFN-induced cytokines and chemokines | Attracts and activates T cells |
| T Cell Response | IFN-responsive T cells | Interferon-gamma, additional cytokines | Amplifies inflammatory signaling to iMSCs |
| Cytokine Networks | Multiple niche cells | TNFα, IL-6, IL-1β [20] [73] | Sustains chronic inflammation, suppresses normal hematopoiesis |
This feed-forward inflammatory loop becomes increasingly dominant as disease progresses from CHIP to MDS, replacing the BM's regenerative architecture with a self-sustaining inflammatory environment that suppresses healthy blood formation and promotes vascular remodeling [72] [75]. Surprisingly, research using the computational tool SpliceUp revealed that mutated hematopoietic cells themselves may not be the primary instigators of this inflammation, highlighting the importance of stromal-immune crosstalk in niche reprogramming [19] [74].
Quantitative Assessment of Microenvironmental Alterations
Multimodal assessment of the bone marrow microenvironment revealed quantitative changes across cellular and molecular dimensions during disease progression.
Table 3: Quantitative Microenvironmental Alterations in CHIP and MDS
| Parameter | Control (n=35) | CHIP (n=17) | MDS (n=32) | Measurement Method | Statistical Significance |
|---|---|---|---|---|---|
| iMSC Frequency | Rare/absent | Emerging population | Expanded population | scRNA-seq, imaging | p<0.01 in MDS vs Control [72] |
| CXCL12 Expression | Normal | Reduced | Significantly reduced | RNA sequencing, proteomics | p<0.001 in MDS vs Control [72] |
| B Cell Populations | Normal | - | Significantly decreased | Immunophenotyping, bulk gene expression | FDR<0.05 [72] |
| TNFα Signaling | Baseline | Elevated | Significantly elevated | GSEA of bulk RNA | FDR<0.05 [72] |
| IFNα Response | Baseline | Elevated | Significantly elevated | GSEA of bulk RNA | FDR<0.05 [72] |
| BM Vasculature | Normal | - | Expanded | Imaging, angiogenic potential assays | p<0.05 [72] |
Bulk gene expression profiling of BM mononuclear cells using NanoString nCounter panels targeting 773 immune-related and 730 cancer inflammation-associated genes identified 123 significantly altered genes between MDS and controls (FDR<0.05), with majority being downregulated in MDS BM [72]. Gene set enrichment analysis (GSEA) revealed upregulation of TNFα and IFNα pathways in MDS, indicating a pro-inflammatory environment, while T cell-related processes were downregulated despite preserved T cell numbers, suggesting functional alteration rather than numeric depletion [72].
Experimental Models and Methodologies
Key Experimental Approaches for Niche Analysis
The following experimental workflows enable comprehensive characterization of the inflammatory bone marrow niche.
Detailed Methodological Protocols
Single-Cell RNA Sequencing of Human Bone Marrow
Sample Processing: Bone marrow mononuclear cells (BMMCs) are isolated from fresh human BM aspirates using density gradient centrifugation (Ficoll-Paque). Cells are resuspended in PBS with 0.04% BSA at a concentration of 1,000-1,200 cells/μL [72].
Library Preparation: Single-cell suspensions are loaded on Chromium Next GEM chips (10x Genomics) to target recovery of 10,000 cells per sample. cDNA amplification is performed following the Chromium Single Cell 3' Reagent Kits v3.1 protocol [72] [19].
Sequencing and Analysis: Libraries are sequenced on Illumina NovaSeq platforms. Raw sequencing data is processed using Cell Ranger pipeline. Downstream analysis includes Seurat for clustering, CellPhoneDB for cell-cell communication inference, and SCENIC for regulatory network analysis [72] [19].
Primary BM HSPC-MSC Co-culture Assays
Stromal Cell Isolation: Primary human MSCs are isolated from BM aspirates by plastic adherence and expanded in MesenCult proliferation medium. Cells are used at passages 2-4 for all experiments [72].
HSPC Co-culture: CD34+ HSPCs are isolated from patient BM using magnetic-activated cell sorting (MACS). For direct co-culture, 5×10⁴ HSPCs are seeded onto confluent MSC layers in StemSpan serum-free medium with cytokines (SCF, TPO, FLT3-L) [72].
Functional Assessment: After 7-10 days of co-culture, hematopoietic output is quantified by flow cytometry for progenitor populations (CD34+CD38-), differentiated cells (CD14+, CD15+, CD19+), and apoptosis (Annexin V). Conditioned media is collected for cytokine profiling via Luminex assay [72].
Computational Tool: SpliceUp for Mutant Cell Identification
A key innovation in distinguishing cell-intrinsic versus microenvironmental effects is the SpliceUp computational tool, which identifies mutated cells within single-cell RNA sequencing data based on aberrant RNA-splicing patterns rather than DNA sequencing [19] [74]. This approach enabled researchers to determine that the inflammatory reprogramming of the niche was not directly attributable to mutant hematopoietic cells, underscoring the autonomy of microenvironmental inflammation in disease pathogenesis [74] [75].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for iMSC and Niche Studies
| Reagent/Category | Specific Examples | Research Application | Functional Role |
|---|---|---|---|
| Single-Cell RNA-seq Kits | Chromium Single Cell 3' Reagent Kits (10x Genomics) | Cell type identification, transcriptional profiling | Comprehensive cellular taxonomy of BM niche |
| Cell Culture Media | MesenCult, StemSpan | Primary MSC and HSPC culture | Maintenance of primary niche cells ex vivo |
| Cell Separation Kits | CD34+ selection kits (MACS) | HSPC isolation from BM | Purification of hematopoietic populations |
| Cytokine Panels | Luminex cytokine arrays | inflammatory secretome analysis | Quantification of inflammatory mediators |
| Spatial Transcriptomics | Visium Spatial Gene Expression | Anatomical niche mapping | Correlation of cellular position and function |
| Computational Tools | SpliceUp, CellPhoneDB | Mutant cell identification, interaction networks | Analysis of cell-cell communication |
Therapeutic Implications and Future Directions
The identification of iMSCs and their role in sustaining a feed-forward inflammatory loop opens new avenues for therapeutic intervention. Rather than targeting mutant hematopoietic cells alone, strategies that disrupt the inflammatory niche may prevent progression from CHIP to MDS or AML [19] [74]. Potential approaches include:
- Anti-inflammatory agents to attenuate the chronic inflammatory signaling that sustains iMSCs [75]
- Interferon-modulating therapies to break the feed-forward loop between iMSCs and T cells [74]
- Stromal-targeted treatments that specifically eliminate iMSCs or reprogram them to a homeostatic state [72]
- Combination therapies that target both malignant clones and their supportive niche [73]
The distinct molecular signatures of iMSCs and interferon-responsive T cells may also serve as biomarkers for early risk stratification, identifying individuals with CHIP who are at highest risk of progression to overt malignancy [19] [75]. Furthermore, understanding the "memory" of the niche following disease has important implications for stem cell transplantation, where residual inflammatory programming might affect engraftment and reconstitution [74] [21].
Inflammatory stromal cells (iMSCs) are central mediators of bone marrow niche dysfunction in CHIP and MDS. Through feed-forward interactions with immune cells, particularly IFN-responsive T cells, iMSCs create a self-sustaining inflammatory environment that suppresses normal hematopoiesis while promoting clonal expansion. This niche remodeling begins early in disease pathogenesis and becomes increasingly dominant as disease progresses. Targeting these inflammatory circuits represents a promising strategy for intercepting malignant progression before overt leukemia develops, shifting the therapeutic paradigm from reactive to preventive approaches in myeloid malignancies.
Vascular Remodeling and Loss of Supportive Signals like CXCL12
The bone marrow (BM) microenvironment, or niche, is a sophisticated ecosystem essential for the maintenance and function of hematopoietic stem cells (HSCs). This whitepaper delves into the critical process of vascular remodeling within the BM niche, with a particular focus on the loss of supportive chemokine signals such as CXCL12. Driven by factors including aging and inflammation, this remodeling significantly disrupts hematopoietic support, contributes to immune defects, and can foster the progression of pre-malignant conditions. We synthesize recent human and murine single-cell studies to detail the molecular and cellular mechanisms involved, present quantitative data on niche alterations, outline key experimental methodologies for its investigation, and discuss emerging therapeutic implications for targeting the niche in hematological disorders.
The bone marrow (BM) niche is a specialized microenvironment that provides both structural and biochemical signals to regulate hematopoietic stem cell (HSC) function, including their self-renewal, differentiation, and retention [8]. This complex regulatory network involves a diverse array of cellular components, such as mesenchymal stromal cells (MSCs), endothelial cells (ECs), osteoblasts, and immune cells, which interact with HSCs through direct contact and the secretion of factors [76] [8].
A critical chemokine within this niche is CXCL12 (also known as SDF-1), produced by various stromal cells. Its primary receptor, CXCR4, is expressed on HSCs. The CXCL12/CXCR4 axis is instrumental for HSC homing to the BM, their retention in specific niche locations, and the maintenance of their quiescence [77]. Recent research has illuminated that the structural and functional integrity of this vascular niche is not static. It undergoes significant remodeling, particularly during aging and in response to chronic inflammation, leading to a detrimental loss of these crucial supportive signals [78] [19] [72].
Mechanisms of Vascular Niche Remodeling and CXCL12 Loss
Vascular remodeling in the BM is characterized by quantitative and qualitative changes in niche cells, driven by inflammatory signals that disrupt the normal supportive environment for HSCs.
Emergence of Inflammatory Stromal Cells
A pivotal change in the remodeling niche is the functional alteration of mesenchymal stromal cells. Single-cell RNA sequencing studies of human BM have identified a distinct population of inflammatory mesenchymal stromal cells (iMSCs) that arise in conditions like clonal hematopoiesis (CHIP) and become more prevalent in myelodysplastic syndromes (MDS) [19] [72].
Unlike supportive MSCs that express high levels of CXCL12 and stem cell factor (SCF), iMSCs exhibit a markedly different transcriptome. They downregulate key hematopoietic support genes such as CXCL12 and KITLG (which encodes SCF) and instead upregulate a pro-inflammatory signature [78] [72]. This signature includes genes encoding chemokines like CXCL2 and CCL2, which attract immune cells, and transcription factors like CEBPB and components of the AP-1 complex (FOSB, JUND), which help sustain the inflammatory state [78]. This shift represents a fundamental change in the stromal landscape from supportive to inflammatory.
Cellular Drivers and Inflammatory Loops
The transformation of the niche is reinforced by a feed-forward inflammatory loop involving immune cells. The iMSCs release large amounts of interferon-induced cytokines and chemokines, which attract and activate interferon-responsive T cells [19] [72]. These T cells, in turn, produce signals that further amplify the inflammatory activation of stromal cells, creating a self-sustaining cycle [72].
This chronic inflammatory microenvironment actively suppresses healthy hematopoiesis. Furthermore, research indicates that in MDS, the mutated hematopoietic stem and progenitor cells (HSPCs) themselves often fail to trigger stromal cells to produce CXCL12, exacerbating the loss of this critical retention signal [19]. Imaging studies have revealed that vascular remodeling is also associated with expanded BM vasculature, coinciding with iMSCs retaining some angiogenic potential even as they lose hematopoietic support function [72].
The following diagram illustrates the sequential process of this inflammatory remodeling in the bone marrow niche.
Figure 1. Inflammatory Remodeling Cascade in the BM Niche. This diagram illustrates the key steps from initial trigger to functional decline, highlighting the self-reinforcing inflammatory loop at its core.
Consequences for Hematopoiesis and Immune Function
The remodeling of the vascular niche and the specific loss of CXCL12 have profound and diverse consequences for blood cell production and immune competence.
- HSC Maintenance and Localization: CXCL12 is a key chemoattractant for HSCs. Studies in conditional knockout mice show that deletion of
Cxcl12from mesenchymal progenitors (usingPrx1-Cre) reduces normal HSC numbers, demonstrating its role in maintenance [79]. Furthermore, research indicates that HSCs actively migrate to remain within range of CXCL12-producing CAR cells within the BM [80]. - Lineage Output and Immune Defects: The loss of CXCL12-supportive niches disproportionately affects lymphopoiesis. Deletion of
Cxcl12from nearly all CAR cells (usingEbf3-CreERT2) markedly reduces the ability of HSCs to generate B cell progenitors, even upon transplantation into a wild-type mouse, suggesting a role in maintaining lymphoid-biased HSCs [80]. This aligns with human BM data from MDS patients showing a pronounced loss of B cells and downregulation of T cell-related genes [72]. - Disease Progression and Therapy Resistance: In the context of chronic myeloid leukemia (CML), deletion of
Cxcl12from MSCs paradoxically promotes leukemic stem cell (LSC) expansion by increasing their self-renewing divisions, potentially through enhanced Ezh2 activity. This indicates that CXCL12 plays a role in maintaining LSC quiescence. Consequently, targeting these interactions can sensitize LSCs to tyrosine kinase inhibitor (TKI) treatment [79].
The table below summarizes key quantitative findings from recent studies on the loss of supportive signals.
Table 1: Quantitative Data on Niche Remodeling and Loss of Supportive Signals
| Parameter Measured | Experimental Context | Observation | Citation |
|---|---|---|---|
| CXCL12 Expression | Human BM: old vs. young individuals | Downregulation in MSC1 population | [78] |
| KITLG (SCF) Expression | Human BM: old vs. young individuals | Downregulation in MSC1 population | [78] |
| Inflammatory Gene Signature | Human BM: iMSCs in CHIP/MDS | Upregulation of CXCL2, CCL2, CEBPB, AP-1 complex genes (FOSB, JUND) |
[78] [72] |
| B Cell Progenitor Production | Mouse: CXCL12 deletion from CAR cells | Markedly reduced ability of HSCs to generate B cell progenitors | [80] |
| LSC Response | Mouse CML model: CXCL12 deletion from MSCs | Increased self-renewing divisions of LSCs; enhanced elimination by TKI treatment | [79] |
Experimental Approaches for Investigating Niche Remodeling
Cutting-edge molecular and cellular techniques are required to dissect the complex changes in the bone marrow microenvironment.
Single-Cell RNA Sequencing (scRNA-seq)
Purpose: To unbiasedly characterize the cellular composition of the BM niche and identify novel cell populations, such as iMSCs, and their transcriptomic signatures [78] [72].
Detailed Protocol:
- Sample Preparation: Obtain human BM aspirates or murine BM. Digest tissue with collagenase and DNase I to create a single-cell suspension [78].
- Cell Enrichment: Deplete CD45+ hematopoietic cells using antibody cocktails (e.g., RosetteSep) to enrich for rare stromal cells. Further enrichment can be achieved using fluorescence-activated cell sorting (FACS) to isolate live (7AAD-), nucleated cells lacking hematopoietic (CD45), erythroid (CD235a, CD71), and plasma cell (SLAMF7) markers [78].
- Library Preparation & Sequencing: Use a platform like 10x Genomics for 3’-end capture and library generation. Sequence the libraries on an Illumina sequencer to a sufficient depth.
- Bioinformatic Analysis: Process raw data using tools like Cell Ranger. Perform downstream analysis with Seurat or Scanpy for clustering, differential expression analysis (identifying markers for iMSCs), and trajectory inference (pseudotime) [78] [72].
Functional Co-culture Assays
Purpose: To directly test the functional capacity of niche cells to support HSPCs and to dissect the molecular mechanisms of this support [72].
Detailed Protocol:
- Stromal Cell Layer: Isolate primary BM MSCs from mouse models (e.g., conditional knockouts) or human donors. Culture them to establish a confluent layer.
- HSPC Co-culture: Isolate HSPCs (e.g., Lin-Sca-1+c-Kit+ [LSK] cells from mice or CD34+ cells from human BM) via FACS. Seed these HSPCs onto the stromal layer in a cytokine-free medium to assess the inherent supportive capacity of the niche cells.
- Intervention (Optional): Introduce neutralizing antibodies against CXCL12 or small-molecule inhibitors (e.g., AMD3100 for CXCR4) to the co-culture to block specific pathways [79].
- Readout: After several days, assay the output. This can include:
- Flow Cytometry: Quantifying the number and phenotype of recovered HSPCs or differentiated progeny.
- Colony-Forming Unit (CFU) Assay: Plating harvested cells in methylcellulose to measure progenitor frequency and lineage potential.
- Phospho-Flow Cytometry: To measure signaling activity (e.g., pSTAT5 in response to IL-7) in HSPCs after co-culture [77].
Lineage Tracing and Conditional Gene Targeting
Purpose: To define the in vivo requirement of a specific factor (e.g., CXCL12) from a defined cellular source (e.g., MSCs vs. endothelial cells) [79] [80].
Detailed Protocol:
- Mouse Model Generation: Cross mice carrying loxP-flanked ("floxed") alleles of the target gene (e.g.,
Cxcl12f/f) with Cre-recombinase driver lines specific to niche cells. Common lines include: - Validation: Confirm efficient gene deletion and specificity using reporter mice (e.g., Rosa26-tdTomato) and RT-qPCR on FACS-sorted niche populations [79] [80].
- Phenotypic Analysis: Analyze the hematopoietic system of the conditional knockout mice for changes in HSC number (via flow cytometry), lineage output (via blood counts and progenitor assays), and response to stressors like transplantation or 5-fluorouracil treatment [79] [80].
The following workflow diagram maps the application of these key methodologies in a typical research pipeline.
Figure 2. Experimental Workflow for Niche Investigation. This diagram shows the cyclical and iterative research process, combining in vivo models, ex vivo profiling, and functional testing.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Research Reagents for Investigating the BM Niche
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
Conditional Knockout Mice (Cxcl12f/f, LepR-Cre, Prx1-Cre) |
To dissect the cell-type-specific function of genes within the complex niche in vivo. | Determining that CXCL12 deletion from MSCs, but not endothelial cells, promotes CML LSC expansion [79]. |
| CXCR4 Inhibitor (AMD3100 / Plerixafor) | Small molecule antagonist of CXCR4; blocks CXCL12/CXCR4 signaling. | Mobilizing HSCs from the niche into peripheral blood; testing dependency of HSC retention on this axis in co-culture [79]. |
| Fluorescence-Activated Cell Sorting (FACS) Antibodies | Isolation of highly pure populations of niche and hematopoietic cells for downstream analysis. | Isolving CD45- Ter119- CD31- stromal cells or LSK HSCs from mouse BM for transcriptomics or transplantation [78] [80]. |
| Single-Cell RNA Sequencing Platforms | Unbiased transcriptional profiling of all cell types in the BM microenvironment. | Identifying novel inflammatory MSC (iMSC) populations in human CHIP and MDS bone marrow [78] [72]. |
| Neutralizing Antibodies (anti-CXCL12, anti-IL-7) | To block the function of specific soluble factors in experimental systems. | Assessing the contribution of specific cytokines to HSPC support in co-culture assays [77]. |
Therapeutic Implications and Future Directions
Understanding vascular niche remodeling opens novel avenues for therapeutic intervention. The identification of iMSCs and inflammatory T-cell loops provides a new cellular target for treatments aimed at intercepting pre-malignant conditions like CHIP before they progress to MDS or AML [19] [72]. Strategies could include anti-inflammatory agents or interferon-modulating drugs to suppress the deleterious iMSC program and restore a more supportive microenvironment [19].
Furthermore, targeting the CXCL12/CXCR4 axis remains a promising strategy. The CML study suggests that disrupting specific LSC-niche interactions could overcome therapy resistance and lead to more effective eradication of leukemic stem cells [79]. Finally, as niche remodeling creates a "memory" of disease, understanding this process is crucial for improving outcomes in stem cell transplantation, potentially through pre-transplant conditioning of the niche [19].
Future work will require longitudinal studies to track niche evolution over time and the development of advanced 3D models that more faithfully recapitulate the human BM microenvironment to test these emerging therapeutic concepts [8] [81].
The bone marrow (BM) microenvironment, or niche, is a highly specialized and dynamic structure that normally regulates the balance between quiescence, self-renewal, and differentiation of hematopoietic stem cells (HSCs). This intricate microenvironment consists of diverse cellular components, extracellular matrix (ECM), and signaling molecules that interact to maintain HSC function [8]. However, this very same protective environment can be hijacked by disseminated tumor cells (DTCs) from various cancers, including breast cancer, prostate cancer, and multiple myeloma, providing them with a sanctuary where they can enter a dormant state and evade therapeutic interventions [82] [83]. Cancer cell dormancy in the bone microenvironment presents a major obstacle to curative therapy across multiple cancer types, enabling therapy evasion and later reactivation to cause disease relapse years or even decades after initial treatment [82]. The bone marrow recruits cancer cells via chemoattractants normally involved in healthy hematopoiesis, particularly the CXCL12-CXCR4 chemokine axis which recruits and retains target cells in the bone marrow [82]. Understanding how the HSC niche protects dormant cancer cells and confers drug resistance is crucial for developing novel therapeutic strategies to prevent metastatic relapse.
Cellular and Molecular Architecture of the Dormancy-Inducing Niche
Key Cellular Components of the Pro-Dormancy Niche
The bone marrow niche contains specific cellular components that create specialized pro-dormancy microenvironments. Dormant cancer cells typically engage with the endosteal and/or perivascular niches when they arrive in the bone marrow, where they can be maintained in a dormant state for extended periods [82]. The cellular architecture of these niches includes:
Bone Lining Cells and Osteoblasts: These cells form the endosteal niche and have been consistently implicated in promoting and maintaining dormancy across different cancer types [82]. Dormant cells have been shown to reside near type I collagen-expressing osteoblasts and osteopontin-positive cells on the endosteal surface [82]. Osteoblasts can protect cancer cells from oxidative damage and hypoxia, creating a favorable environment for dormancy [82] [8].
Nestin+ NG2+ Mesenchymal Stromal Cells (MSCs): These specific perivascular stromal cells have been identified as critical components of the pro-dormancy niche, particularly for breast cancer DTCs [82]. These MSCs shape the bone marrow microenvironment by secreting regulatory factors and have been shown to promote cellular dormancy through various signaling pathways [82] [84] [8].
Endothelial Cells: These cells form the vascular niche and regulate cell migration, maintenance, and activation. The perivascular niche is protective to breast DTCs in the bone marrow, providing integrin-mediated resistance to chemotherapy [82]. Endothelial cells support stem cell homeostasis and can mobilize cells in response to stress or injury [8].
Megakaryocytes: These cells are an important component of the HSC niche and are thought to regulate quiescence by secreting various factors including CXCL4 [20]. Analysis of the spatial relationship between HSCs and megakaryocytes has shown that HSCs are significantly closer to megakaryocytes in the niche, supporting a functional relationship that may extend to cancer cell dormancy [20].
Immune Cells: Macrophages support cell maintenance by secreting cytokines like IL-6 and TGF-β, while specialized nestin+ macrophages help regulate cell retention via MSC interactions [8]. The bone marrow niche also contains regulatory T cells (Tregs) that can establish a survival-promoting niche through direct interactions with resident cells [7].
Molecular Regulators of Dormancy in the Bone Marrow Niche
The cellular components of the niche produce numerous molecular factors that directly regulate cancer cell dormancy. These signaling molecules work in concert to maintain DTCs in a quiescent state while simultaneously promoting their survival and therapy resistance.
Table 1: Key Molecular Regulators of Cancer Cell Dormancy in the Bone Marrow Niche
| Molecular Factor | Cellular Source | Function in Dormancy | Target Cancer Types |
|---|---|---|---|
| CXCL12 (SDF-1) | Stromal and endothelial cells | Regulates DTC retention, homing, and mobilization via CXCR4 | Breast cancer, Prostate cancer, Myeloma [82] [8] |
| GAS6 | Osteoblast lineage cells, Breast cancer cells | Binds to TYRO3 receptor, promotes maintenance of dormancy | Breast cancer, Multiple myeloma [82] |
| TGF-β2 | Bone microenvironment, MSCs | Induces growth arrest, promotes cellular dormancy | Breast cancer, Prostate cancer [82] [84] |
| BMP-7 | Bone stromal cells, NG2+Nestin+ MSCs | Induces dormancy via p38 pathway and upregulation of NDRG1 | Prostate cancer [82] [84] |
| Leukaemia Inhibitory Factor (LIF) | Bone marrow stromal cells | Drives dormancy in bone through STAT3 signaling | Breast cancer [82] [84] |
| All-trans Retinoic Acid (atRA) | Bone microenvironment | Cooperates with intrinsic tumor signals to promote dormancy | Breast cancer [84] |
The balance between proliferation and dormancy is tightly regulated by key signaling pathways within cancer cells. A crucial mechanism involves the ratio of extracellular signal-regulating kinases (ERKs) to p38 mitogen-activated protein kinase (MAPK) [84] [83]. When ERK signaling predominates, cells proliferate, whereas when p38 signaling is elevated, cells enter dormancy. The lower ERK/p38 expression ratio serves as a key indicator of the dormant state in cancer cells [84]. Additionally, proteins encoded by the F-box and WD repeat domain containing 7 (FBXW7) gene regulate mitotic activity by targeting key proteins such as cyclin E and c-Myc for degradation, thereby suppressing proliferation and maintaining quiescence [84].
Diagram 1: Signaling network in the bone marrow niche that induces and maintains cancer cell dormancy. Key niche cells produce molecular factors that activate dormancy pathways in cancer cells through receptor-mediated signaling and p38 MAPK activation, leading to cell cycle arrest and therapy resistance.
Mechanisms of Therapy Resistance and Immune Evasion in Dormant Cancer Cells
Intrinsic and Microenvironment-Mediated Resistance Mechanisms
Dormant cancer cells employ multiple strategies to resist conventional therapies and evade immune surveillance within the bone marrow niche:
Cell Cycle-Mediated Resistance: As dormant cells are non-cycling and arrested in the G0/G1 phase of the cell cycle, they are largely resistant to therapies that target rapidly dividing cells, such as conventional chemotherapy [82] [84]. This quiescent state enables them to survive treatment and potentially cause disease relapse later.
Autophagy and Metabolic Adaptation: Studies have identified autophagy as a key supporting mechanism for dormancy, with a degree of built-in redundancy [82]. This cellular self-digestion process helps dormant cells survive stress and limited nutrient availability in the bone marrow microenvironment. These advances have led to ongoing clinical trials testing autophagy inhibitors like hydroxychloroquine as potential dormancy-targeting therapies [82].
Stemness and Plasticity: Dormancy has been linked to enhanced stemness and increased resistance to therapies [84]. Cancer stem cells (CSCs) and dormant cells share traits, with some CSCs being able to enter a dormant state. Epigenetic modifications play a crucial role in this process, with studies showing that endocrine therapies can induce epigenetic modifications that promote dormancy induction [82].
Adhesion-Mediated Resistance: The perivascular niche provides integrin-mediated resistance to chemotherapy for DTCs in the bone marrow [82]. Targeting endothelial-derived von Willebrand factor and vascular cell adhesion molecule 1 (VCAM1) has been shown to sensitize mice to chemotherapy and prevent bone metastases [82].
Immune Evasion Strategies
The bone marrow niche offers protection from immune surveillance through multiple mechanisms:
Immunosuppressive Secretome: The bone marrow microenvironment produces various immunosuppressive factors that can inhibit anti-tumor immune responses. Mesenchymal stem cells (MSCs) support HSCs by secreting regulatory factors that maintain quiescence and retention, creating an overall immunosuppressive environment [8].
Regulatory T Cell Recruitment: The bone marrow niche contains regulatory T cells (Tregs) that can establish a survival-promoting niche for aged HSCs through direct interactions [7]. These Tregs likely contribute to the immune privilege of dormant cancer cells within the niche.
Altered Antigen Presentation: Dormant cancer cells may downregulate antigen presentation machinery, making them less visible to immune detection. While not explicitly detailed in the search results, this is a common mechanism of immune evasion in cancer that likely applies to dormant cells in the bone marrow niche.
Table 2: Therapy Resistance Mechanisms of Dormant Cancer Cells in the Bone Marrow Niche
| Resistance Mechanism | Key Mediators | Therapeutic Implications |
|---|---|---|
| Cell Cycle Arrest | p21, p27, p38 MAPK | Resistance to cell cycle-targeting chemotherapies [82] [84] |
| Autophagy | Autophagy-related proteins | Clinical trials with hydroxychloroquine (autophagy inhibitor) [82] |
| Enhanced Stemness | Epigenetic modifiers, TWIST1, CSC markers | Targeting epigenetic regulators; combination therapies [84] |
| Adhesion-Mediated Resistance | Integrins, VCAM1, von Willebrand factor | Targeting adhesion molecules to sensitize to chemotherapy [82] |
| Immune Evasion | Regulatory T cells, Immunosuppressive cytokines | Immune checkpoint inhibitors; targeting niche immune components [7] [8] |
Experimental Models and Methodologies for Studying Dormancy
Advanced 3D Biomimetic Models of the Bone Marrow Niche
Recent advances have shifted from conventional two-dimensional (2D) culture systems toward three-dimensional (3D) biomimetic models through the development of artificial bone marrow niches [7]. The integration of cutting-edge platforms—such as 3D printing, organoids, and bone marrow-on-a-chip—has enabled applications including in vitro HSC expansion, lineage-specific differentiation, disease modeling, and high-throughput drug screening [7]. These models aim to recreate the physiological extracellular matrix organization to support long-term haematopoietic stem cells and, by extension, study cancer cell dormancy in a more physiologically relevant context.
One particularly innovative approach reported in Nature Communications uses bioengineered niches that recreate physiological extracellular matrix organisation using soft collagen type-I hydrogels to drive nestin expression in perivascular stromal cells (PerSCs) [39]. When CD34+ve HSCs were added to these bioengineered niches comprising nestin/HIF-1α expressing PerSCs, long-term HSC numbers were maintained with normal clonal and in vivo reconstitution potential, without media supplementation [39]. This system provides a valuable platform for studying cancer cell dormancy mechanisms and testing potential therapeutic interventions.
Detailed Experimental Protocol: Bioengineered Niche for Dormancy Studies
Protocol Title: Establishment of a Bioengineered Bone Marrow Niche to Study Cancer Cell Dormancy
Background: This protocol describes the creation of a bioengineered LT-HSC maintenance niche that recreates physiological extracellular matrix organisation, using soft collagen type-I hydrogels to drive nestin expression in perivascular stromal cells (PerSCs) [39]. The system can be adapted to study cancer cell dormancy mechanisms and test therapeutic interventions.
Materials and Equipment:
- Poly(ethyl acrylate) (PEA) or poly(methyl acrylate) (PMA) surfaces
- Fibronectin (FN) from human plasma
- Soft collagen type-I hydrogels (Young's modulus: 1–104 Pa)
- Perivascular stromal cells (PerSCs) or Mesenchymal stromal cells (MSCs)
- Cancer cells of interest (e.g., breast cancer, prostate cancer cells)
- Hypoxia chamber or hypoxia-inducible factors
- Antibodies for detection: anti-nestin, anti-HIF-1α, anti-fibronectin domains
Procedure:
- Surface Preparation: Coat culture surfaces with PEA, which leads to spontaneous unfolding of fibronectin (FN), or with PMA as a control, on which FN is adsorbed and maintained in a globular conformation [39].
- Hydrogel Fabrication: Prepare soft collagen type-I hydrogels with mechanical properties mimicking the bone marrow niche (Young's modulus: 1–104 Pa) [39] [7].
- Stromal Cell Seeding: Seed PerSCs or MSCs onto the prepared surfaces and hydrogels. Culture under hypoxic conditions (1-5% O₂) to mimic bone marrow oxygenation [39].
- Niche Characterization: Verify nestin and HIF-1α expression in stromal cells using immunostaining or Western blotting. Confirm the enhanced availability of FN domains when absorbed on PEA compared to PMA [39].
- Cancer Cell Introduction: Add cancer cells of interest to the established bioengineered niches. Use appropriate cancer cell lines such as breast cancer (MDA-MB-231) or prostate cancer cells [39].
- Dormancy Assessment: Monitor cancer cell dormancy using the following approaches:
- Cell cycle analysis (Ki-67 negative, p27 positive)
- ERK/p38 MAPK ratio quantification
- Resistance to chemotherapeutic agents
- Long-term culture initiating cell (LTC-IC) assays [39]
Applications:
- Study dormancy induction and maintenance mechanisms
- Test dormancy-breaking factors
- Screen potential therapeutic compounds targeting dormant cells
- Investigate cancer cell-niche interactions
- Perform genetic manipulation of dormant cells (e.g., CRISPR editing) [39]
Diagram 2: Experimental workflow for establishing a bioengineered bone marrow niche to study cancer cell dormancy. The protocol involves sequential steps from surface preparation to therapeutic testing, creating a physiologically relevant system for dormancy research.
The Scientist's Toolkit: Essential Research Reagents for Dormancy Studies
Table 3: Key Research Reagents for Investigating Cancer Cell Dormancy in the Bone Marrow Niche
| Research Tool | Specific Examples | Application/Function |
|---|---|---|
| Biomimetic Hydrogels | Soft collagen type-I hydrogels (1-104 Pa) | Recreate mechanical properties of bone marrow niche; induce nestin expression in stromal cells [39] [7] |
| Specialized Polymers | Poly(ethyl acrylate) (PEA), Poly(methyl acrylate) (PMA) | Control fibronectin conformation; enhance availability of integrin and growth factor binding domains [39] |
| Stromal Cell Markers | Anti-nestin, Anti-NG2, Anti-HIF-1α antibodies | Identify and characterize pro-dormancy niche cells [82] [39] |
| Dormancy Markers | Ki-67 (negative), p27, p21, phospho-p38, ERK/p38 ratio | Detect and quantify dormant cell state; distinguish from proliferating cells [82] [84] [83] |
| Cytokines & Growth Factors | Recombinant TGF-β2, BMP-7, GAS6, CXCL12 | Investigate dormancy induction and maintenance mechanisms [82] [84] |
| Signaling Inhibitors | p38 inhibitors, Autophagy inhibitors (Hydroxychloroquine), ERK activators | Probe molecular mechanisms; potential therapeutic interventions [82] [84] |
| 3D Culture Systems | Bone marrow-on-a-chip, Organoids, 3D bioprinted scaffolds | Model complex niche interactions; high-throughput drug screening [7] [39] |
Therapeutic Strategies and Future Directions
Targeting Dormancy Mechanisms for Therapeutic Intervention
Several promising therapeutic approaches are emerging that target different aspects of cancer cell dormancy in the bone marrow niche:
Autophagy Inhibition: With autophagy identified as a key supporting mechanism for dormancy, autophagy inhibitors such as hydroxychloroquine are being investigated in clinical trials as potential dormancy-targeting therapies [82]. These approaches aim to disrupt the survival mechanisms that allow dormant cells to persist in the bone marrow niche.
Combination Therapies: Given that dormant cells are resistant to conventional therapies, combination approaches that target both proliferating cells and dormant populations hold promise [84]. These might include chemotherapy combined with dormancy-targeting agents or drugs that prevent reactivation.
Niche-Directed Therapies: Instead of directly targeting cancer cells, these approaches modify the bone marrow niche to make it less supportive of dormancy. This could involve targeting key niche components such as NG2+Nestin+ MSCs or disrupting pro-dormancy signals like TGF-β2 and BMP-7 [82] [84].
Immune-Mediated Approaches: Strategies that enhance immune surveillance against dormant cells show potential. These include using IL-15 to activate NK cells and stimulate interferon-γ production, which can induce tumor cells into dormancy or maintain them in that state [85]. Checkpoint inhibitors might also help the immune system recognize and eliminate dormant cells.
Future Research Directions
The field of cancer dormancy research is rapidly evolving, with several promising directions for future investigation:
Single-Cell Dynamics: A deeper understanding of dormancy dynamics at the single-cell level is needed, including the duration of quiescent periods, frequency of switching between quiescent and cycling states, and potential heterogeneity across different skeletal sites [82].
Spatial Heterogeneity: Investigation into whether different skeletal sites provide distinct pro-dormancy niches, given that different skeletal sites have different niche and blood cell compositions and different responses to stress [82].
Metabolic Dependencies: Further exploration of the metabolic adaptations that allow dormant cells to survive in the bone marrow niche could reveal new therapeutic vulnerabilities [82] [39].
Advanced Model Systems: Continued development of more sophisticated experimental models, including improved bioengineered niches, patient-derived organoids, and humanized mouse models that better recapitulate the human bone marrow microenvironment [7] [39].
The ongoing development of increasingly sophisticated experimental models of the bone marrow niche, combined with single-cell technologies and advanced imaging approaches, promises to accelerate our understanding of the mechanisms governing cancer cell dormancy. This knowledge will be crucial for developing effective therapies to target dormant cells and prevent metastatic relapse, ultimately improving long-term outcomes for cancer patients.
The hematopoietic stem cell (HSC) niche is a specialized bone marrow microenvironment that provides structural and biochemical support to regulate critical HSC behaviors, including quiescence, self-renewal, differentiation, and homing [86] [8]. This dynamic microenvironment consists of a complex network of cellular components, extracellular matrix (ECM) proteins, and signaling molecules that work in concert to maintain hematopoietic homeostasis [7] [8]. The niche not only supports normal hematopoiesis but also plays a significant role in the etiology of various hematological disorders when disrupted [7]. Over the past decade, increased attention has focused on understanding how manipulation of this niche can yield therapeutic benefits, particularly for hematopoietic stem cell transplantation and the treatment of hematological malignancies [87] [88] [89].
The cellular architecture of the HSC niche includes heterologous cell-cell interactions involving osteoblasts, endothelial cells, mesenchymal stromal cells (MSCs), adipocytes, macrophages, and megakaryocytes [7] [8] [9]. These cells regulate HSC function through direct contact and secretion of cytokines, growth factors, and chemokines such as SCF, TPO, CXCL12, and SDF-1 [7] [8]. Beyond biochemical signaling, the niche also provides physical regulation through matrix stiffness, viscoelasticity, topological architecture, and fluid shear stress [7]. The mechanical properties of the niche significantly influence HSC fate decisions, with the endosteal niche exhibiting a relatively rigid matrix (>35 kPa) and the vascular niche characterized by softer matrices (0.3-8 kPa) [7].
Table 1: Major Cellular Components of the HSC Niche
| Cell Type | Primary Functions | Key Signaling Molecules |
|---|---|---|
| Mesenchymal Stem Cells (MSCs/CAR/LepR+ cells) | Major niche component supporting HSC maintenance; differentiates into osteoblasts, adipocytes [9] | CXCL12, SCF, KIT ligand [8] [9] |
| Endothelial Cells | Form vascular niche; regulate HSC migration, maintenance, activation [8] | Notch ligands, VEGF, angiocrine factors [8] |
| Osteoblasts | Maintain endosteal niche; regulate HSC quiescence [8] | Osteopontin, angiopoietin-1, Wnt, BMP [8] |
| Megakaryocytes | Regulate HSC quiescence [7] [20] | CXCL4, TPO, TGF-β [7] |
| Macrophages | Support HSC maintenance; preserve niche homeostasis [8] | IL-6, TGF-β [8] |
| Adipocytes | Negative or positive regulators of HSCs depending on context [9] | Adiponectin, leptin, MCP-1 [35] |
Hematopoietic Stem Cell Mobilization Strategies
Granulocyte Colony-Stimulating Factor (G-CSF) Mechanism
G-CSF represents the cornerstone of clinical HSC mobilization protocols, functioning primarily through attenuation of the bone marrow niche. The mechanism involves remarkable osteoblast "flattening" and reduction of key niche components including osteopontin (OPN), stromal-derived factor-1 (SDF-1), and N-cadherin expression [88]. G-CSF also reduces resident F4/80+ monocytes/macrophages that normally support niche osteoblasts and mesenchymal stromal cells, further contributing to niche attenuation and hematopoietic mobilization [88]. The disruption of these critical retention signals facilitates egress of HSCs from the bone marrow into peripheral blood circulation.
The SDF-1/CXCR4 axis represents a well-established pathway for niche retention, and while G-CSF reduces SDF-1 expression, its mobilization efficacy involves additional complex mechanisms [88]. Research demonstrates that G-CSF-mediated mobilization remains partially effective even in CXCR4 knockout mice, suggesting the involvement of alternative pathways [88]. The synergistic relationship between G-CSF and other mobilizing agents, particularly non-steroidal anti-inflammatory drugs (NSAIDs), highlights the multifactorial nature of niche disruption and HSC release [88].
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) as Mobilizing Agents
NSAIDs have emerged as a novel therapeutic strategy for HSC mobilization, working through distinct but complementary mechanisms to G-CSF. Short-term administration of the NSAID meloxicam demonstrates significant attenuation of the BM niche, including reduced osteoid bone surfaces with an approximately 3-fold increase in quiescent surfaces [88]. Similar to G-CSF, NSAID treatment results in marked reductions in osteopontin (OPN), SDF-1, and N-cadherin expression [88].
The NSAID mobilization mechanism exhibits both similarities and distinctions compared to G-CSF. While both agents attenuate the niche microenvironment, they function through independent pathways, potentially explaining their synergistic mobilization effect when combined [88]. A critical distinction lies in their differential effects on osteopontin: NSAID-mediated OPN reduction is specifically responsible for HSC mobilization, while HPC mobilization appears to be mediated by another mechanism [88]. This intriguing finding suggests the possibility of targeted niche attenuation for specific therapeutic applications.
Table 2: Quantitative Effects of Mobilizing Agents on Bone Marrow Niche Components
| Niche Component | G-CSF Treatment | NSAID (Meloxicam) Treatment |
|---|---|---|
| Osteoblast Morphology | Remarkable "flattening" [88] | Remarkable "flattening" [88] |
| Osteoid Bone Surfaces | Significantly reduced [88] | Significantly reduced (~3-fold increase in quiescent surfaces) [88] |
| SDF-1 Expression | Marked reduction [88] | Marked reduction [88] |
| Osteopontin (OPN) Expression | Marked reduction [88] | Marked reduction [88] |
| N-cadherin Expression | Marked reduction [88] | Marked reduction [88] |
| F4/80+ Monocytes/Macrophages | Reduced [88] | No significant alteration [88] |
| Osteoclast Activity | Slightly elevated [88] | Slightly elevated [88] |
Figure 1: Mechanism of HSC Mobilization by G-CSF and NSAIDs. Both agents attenuate the bone marrow niche through overlapping but distinct pathways, ultimately leading to HSC/HPC mobilization.
Anti-Inflammatory Strategies in Niche Modulation
Inflammation in the Aging HSC Niche
The aging hematopoietic niche undergoes significant functional and structural alterations characterized by chronic low-grade inflammation ("inflammaging"), increased adiposity, and impaired support capacity [20] [35]. Aged immune cells become the main contributors to inflammaging through secretion of inflammatory cytokines such as IL-1β, which creates a vicious cycle of increased HSPC proliferation and clonal expansion that contributes to conditions like Clonal Hematopoiesis of Indeterminate Potential (CHIP) [20]. The chemokine Ccl5 (RANTES) is notably enriched in the aged microenvironment and exposure of young HSCs to Ccl5 induces the same myeloid bias observed in aged HSCs [20].
The most striking morphological change in the aged human bone marrow is the development of yellow marrow, which consists predominantly of adipocytes and increases from approximately 42% in young individuals to 71% by age 80 [9]. This adipocyte expansion derives from CAR/LepR+ cells and creates a profoundly different regulatory environment through altered secretion of adipokines such as adiponectin, which exhibits both pro-inflammatory and anti-inflammatory functions [9] [35]. These age-related changes create a microenvironment that promotes premature immune cell activation and alters normal differentiation patterns, ultimately contributing to immunosenescence [35].
NSAIDs in Cancer Prevention and Niche Modulation
Epidemiological evidence strongly supports the role of NSAIDs, particularly aspirin, in reducing cancer incidence and mortality. Long-term NSAID use associates with reduction in incidence and mortality across various cancers, particularly colorectal cancer [90]. In 2016, the US Preventive Services Task Force formally recommended low-dose aspirin for primary prevention of colorectal cancer in adults aged 50-59 years [90]. Similar risk reductions have been documented for gastric, endometrial, breast, esophageal, and liver cancers [90].
The anti-tumor mechanisms of NSAIDs extend beyond cyclooxygenase inhibition to include modulation of the tumor microenvironment. NSAIDs target multiple inflammatory pathways implicated in carcinogenesis, including NF-κB and STAT3 signaling, which control the expression of numerous carcinogenic genes that enhance cancer cell survival, proliferation, invasion, and metastasis [90]. Additionally, NSAIDs may impact the bone marrow niche by reducing the production of pro-inflammatory cytokines such as TNFα, type I and type II interferon, and other inflammatory mediators [90].
Experimental Models and Methodologies
In Vitro HSC Niche Models
Advanced in vitro models have been developed to replicate the complex bone marrow microenvironment for research applications. These systems have evolved from conventional two-dimensional (2D) culture systems toward three-dimensional (3D) biomimetic models through the development of artificial bone marrow niches [7]. Integration of cutting-edge platforms such as 3D printing, organoids, and bone marrow-on-a-chip has enabled applications including in vitro HSC expansion, lineage-specific differentiation, disease modeling, and high-throughput drug screening [7].
A representative protocol for establishing in vitro HSC niche models utilizes freshly isolated bone marrow cells from young (2-3 months) and aged (24 months) C57BL/6JRccHsd mice [35]. Cells are resuspended in complete long-term culture medium (MyeloCult M5300 medium with freshly prepared hydrocortisone at 1×10⁻⁶ M final concentration) and seeded at 1.1×10⁶ cells/cm² in cell culture flasks [35]. Cultures are maintained in a humidified CO₂ incubator (33°C, 5% CO₂) for 4 weeks with half-medium replacement weekly, after which adherent cells are washed and exposed to supernatant media for 48 hours to generate conditioned media representing young versus old niche environments [35].
Heterochronic Transplantation Models
Heterochronic transplantation provides a powerful approach to test the influence of aged versus young recipient niches on HSC function. These experiments demonstrated that donor HSC engraftment is reduced if the recipient niche is aged, while conversely, the young niche can rejuvenate aged donor HSCs [20]. Transplantation of aged HSCs and progenitors into young recipients can partly reverse the aging phenotype, while young HSCs and progenitors can adopt an aged phenotype when transplanted into aged recipients [20]. This provides strong evidence that the bone marrow microenvironment significantly impacts HSC function throughout the lifespan.
In one study, transplantation of hematopoietic progenitor cells into an aged microenvironment produced the characteristic increase in myeloid and decrease in lymphoid cell output associated with aged HSCs [20]. Furthermore, transplant of aged HSCs into Ccl5 knockout recipients helped balance lineage output, with significantly fewer myeloid and more B cells being produced, indicating that modulation of inflammatory signals in the niche can ameliorate age-related hematopoietic dysfunction [20].
Figure 2: Experimental Workflow for In Vitro HSC Niche Modeling. This diagram illustrates the process of establishing and analyzing young versus aged niche environments.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for HSC Niche Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Culture Media | MyeloCult M5300 [35] | Specialized medium for long-term culture of hematopoietic cells |
| Cytokines/Growth Factors | G-CSF, SCF, TPO, CXCL12/SDF-1 [7] [8] | HSC mobilization, maintenance, and differentiation studies |
| Small Molecule Inhibitors | Cdc42 inhibitor [20], NSAIDs (Meloxicam) [88] | Targeting niche signaling pathways, HSC mobilization |
| Cell Isolation | Antibodies for CD34, CD150, CD48, CD41 [9] | Identification and sorting of HSC subpopulations |
| Conditioned Media | From young vs. aged niche models [35] | Studying age-related niche effects on target cells |
| Animal Models | C57BL/6JRccHsd mice [35], CXCR4 KO, OPN KO [88] | In vivo validation of niche mechanisms |
Niche-directed therapies represent a promising frontier in regenerative medicine and hematological disease treatment. The strategic manipulation of the bone marrow microenvironment through mobilizing agents like G-CSF and NSAIDs, coupled with anti-inflammatory approaches, offers significant potential for improving hematopoietic stem cell transplantation outcomes and combating age-related hematopoietic decline. The demonstrated synergy between different classes of mobilizing agents highlights the therapeutic advantage of targeting multiple niche components simultaneously [88].
Future developments in this field will likely focus on increasingly precise temporal and spatial control of niche modulation. As noted by researchers, "successful treatments in regenerative medicine will involve different combinations of factors to target stem cells and niche cells, applied at different times to effect recovery according to the dynamics of stem cell–niche interactions" [87]. Advancements in single-cell technologies, organoid models, and biomimetic scaffolds will further enhance our understanding of niche complexity and enable more targeted therapeutic interventions [7] [86]. The continued elucidation of niche biology, particularly in pathological conditions such as acute myeloid leukemia where the niche facilitates immune escape, will provide new opportunities for combinatorial approaches that prevent tumor evasion mechanisms while minimizing toxicity [89].
The hematopoietic stem cell (HSC) niche represents a highly complex and dynamically regulated bone marrow microenvironment that is indispensable for maintaining lifelong hematopoiesis. This specialized niche not only supports HSC survival, proliferation, and differentiation but also actively shapes the stem cell hierarchy by creating specific microenvironments that confer immune privilege and functional regulation [7]. For researchers and drug development professionals, understanding and accurately replicating this microenvironment has become paramount for advancing both basic research and clinical applications in hematological disorders.
The technical challenges in standardizing culture protocols and translating in vitro findings to clinically relevant outcomes represent significant bottlenecks in the field. Current research aims to bridge the gap between simplified in vitro models and the intricate physiological reality of the bone marrow microenvironment. This whitepaper examines these core challenges within the broader context of HSC niche research and provides a comprehensive technical guide to current methodologies, standardized protocols, and validation frameworks essential for overcoming these translational barriers.
Core Technical Challenges in HSC Niche Modeling
Reconstructing the HSC niche in vitro presents multiple technical hurdles that stem from the inherent complexity of the native bone marrow microenvironment. The table below summarizes the primary challenges and their research implications:
Table 1: Core Technical Challenges in HSC Niche Modeling
| Challenge Category | Specific Technical Hurdles | Impact on Research & Translation |
|---|---|---|
| Microenvironmental Complexity | Multicellular crosstalk between osteoblasts, endothelial cells, mesenchymal cells, and neural components [7] | Simplified models fail to recapitulate native signaling networks and HSC behavior |
| Biophysical Cues | Replicating physiological stiffness gradients (0.3-35 kPa), viscoelasticity, and 3D topological architecture [7] | Altered mechanotransduction leads to dysfunctional HSC fate decisions |
| Biochemical Gradient Formation | Maintaining stable oxygen gradients, cytokine concentrations (SCF, CXCL12, TPO), and calcium ion levels [7] | Loss of HSC quiescence and impaired long-term repopulation capacity |
| Spatial Architecture | Recreating endosteal-perivascular niche compartmentalization and vascular networks [91] [7] | Defective HSC homing, engraftment, and lineage-specific differentiation |
| Model Standardization | Inter-protocol variability in biomaterials, cell sources, and culture conditions [7] [28] | Poor reproducibility between laboratories hinders clinical translation |
A critical aspect of the biochemical challenge involves the cellular source of signaling molecules. Research demonstrates that the functional output of a niche signaling pathway is dictated by both the ligand and its cellular origin. For instance, SDF-1 production by osteolineage cells preferentially regulates multipotent progenitors (MPP) and common lymphoid progenitors (CLP) retention, while endothelial-derived SDF-1 is crucial for HSC maintenance and quiescence [7]. Similarly, although SCF is secreted by multiple stromal cells, HSC maintenance specifically relies on the endothelial source [7]. This cellular specificity adds layers of complexity to in vitro niche reconstruction.
Standardizing Culture Protocols: From 2D to Advanced 3D Systems
Evolution from Conventional to Biomimetic Platforms
Initial in vitro culture of HSCs predominantly relied on cytokine-dependent two-dimensional (2D) systems, but the inherent instability of cytokines and lack of physiological context prompted a shift toward co-culture systems incorporating niche cells [7]. Evidence demonstrates that co-cultured cells enhance HSC maintenance and expansion through paracrine mechanisms, yet the complexity and functional fidelity of these systems remain inferior to the in vivo niche.
Recent advancements have driven a paradigm shift from conventional 2D culture systems toward three-dimensional (3D) biomimetic models through the development of artificial bone marrow niches [7] [28]. The integration of cutting-edge platforms—including 3D printing, organoids, and bone marrow-on-a-chip—has enabled applications spanning in vitro HSC expansion, lineage-specific differentiation, disease modeling, and high-throughput drug screening.
Table 2: Comparison of HSC Culture Platforms
| Culture Platform | Key Components | Applications | Limitations |
|---|---|---|---|
| 2D Co-culture Systems | Stromal feeder layers, cytokine cocktails | Short-term HSC maintenance, preliminary toxicity screening | Limited niche complexity, rapid HSC differentiation |
| Biomimetic Hydrogel Scaffolds | Gelatin methacrylamide (GelMA), hyaluronic acid, collagen matrices | Long-term HSC expansion (weeks), directed differentiation | Batch-to-batch variability in matrix composition |
| Bone Marrow Organoids (BMOs) | Mesenchymal stem cells, endothelial cells, osteoblasts | Disease modeling, studying cell-cell interactions | Challenges in controlling size and reproducibility |
| Bone Marrow-on-a-Chip | Microfluidic channels, endothelialized chambers, mechanical perfusion | Hematopoietic toxicity testing, drug screening | Specialized equipment requirements, technical expertise |
| 3D Bioprinted Niches | Spatial patterning of multiple niche cell types, customized geometries | Personalized medicine applications, complex niche modeling | High cost, limited resolution for microvasculature |
Methodological Framework for 3D HSC Niche Reconstruction
A standardized protocol for constructing 3D bone marrow organoids incorporates the following key methodological steps:
Scaffold Fabrication: Prepare gelatin-hyaluronic acid (Gel-HA) hybrid hydrogels with stiffness optimized between 0.5-2 kPa to mimic physiological perivascular niche mechanics [7].
Stromal Compartment Seeding: Isplicate and expand primary human mesenchymal stem cells (MSCs) from bone marrow aspirates. Seed MSCs at density of 5×10^5 cells/mL in hydrogel scaffold and culture for 7 days to establish stromal network.
Endothelial Network Formation: Isolate human umbilical vein endothelial cells (HUVECs) and inject at density of 1×10^6 cells/mL into pre-established stromal constructs. Culture under angiogenic conditions (50 ng/mL VEGF) for 14 days with medium exchange every 48 hours.
HSC Incorporation and Maintenance: Isolate CD34+ hematopoietic stem and progenitor cells (HSPCs) from umbilical cord blood using immunomagnetic separation. Introduce HSPCs at density of 1×10^4 cells/mL into matured organoids. Maintain in serum-free expansion medium (SFEM) supplemented with defined cytokine cocktail (100 ng/mL SCF, 100 ng/mL TPO, 50 ng/mL FLT3-L) with half-medium changes every 72 hours [92].
Quality Control Metrics: Validate organoid functionality through:
- Flow cytometric analysis of CD34+CD38- HSC population maintenance
- Colony-forming unit (CFU) assays at weekly intervals
- Limiting dilution transplantation assays for in vivo repopulation capacity
This protocol typically enables HSC expansion between 236- to 899-fold over a month when optimized, though significant clone-to-clone variability persists in single cell-initiated cultures [92].
Diagram 1: 3D HSC Niche Reconstruction Workflow
Translating In Vitro Findings: Validation and Clinical Predictive Value
Bridging the In Vitro-In Vivo Gap
A persistent challenge in HSC niche research is the limited predictive value of in vitro models for clinical outcomes. Even with current knowledge and expertise in translational sciences, the success rate of drugs is only 7% in drug development, mainly due to a lack of efficacy in target patient groups [93]. This translational failure underscores the critical need for more physiologically relevant HSC niche models.
Advanced validation frameworks for HSC niche models should incorporate multiple complementary approaches:
Functional Transplantation Assays: The gold standard for validating HSC function remains in vivo repopulation capacity. Limiting dilution transplantation into immunodeficient mice (NSG or NRG strains) with assessment of multilineage engraftment over 16-20 weeks provides critical functional data [92]. Secondary transplantation further evaluates self-renewal capacity.
Multi-omics Integration: Single-cell RNA sequencing (scRNA-seq) of in vitro expanded HSCs enables comparison with native bone marrow HSC transcriptional profiles. Identification of differential expression in key self-renewal regulators (HOXB4, ESAM, ANGPTL, FSTL1, PRDM16) provides molecular validation [92].
Biomarker Correlation: Tracking established HSC biomarkers (ADGRG1, CD34, CD90, CD45RA) across culture duration confirms maintenance of primitive phenotype. Recent scRNA-seq data suggests ADGRG1 may be a potential marker for functional HSCs in ex vivo expanded cells under oxidative stress conditions [92].
Metabolic Profiling: Assessment of glycolytic and oxidative phosphorylation rates provides functional metabolic validation, as HSCs predominantly utilize anaerobic glycolysis rather than mitochondrial oxidative phosphorylation.
Quantitative Framework for Translation
Stochastic modeling approaches offer powerful tools for translating in vitro observations to predicted in vivo behaviors. The HematopoiesisSimulator provides user-friendly capabilities for stochastic simulation and visualization of hematopoietic processes, allowing researchers to conduct virtual experiments and obtain estimates with uncertainty quantification [94] [95]. This computational approach models HSC behavior using three key parameters: λ (mean replication rate), α (mean apoptosis rate), and ν (mean differentiation rate) [94].
Diagram 2: In Vitro to In Vivo Translation Framework
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful HSC niche reconstruction requires carefully selected reagents and platforms that enable accurate mimicry of the native bone marrow microenvironment. The following table details essential research solutions and their applications:
Table 3: Essential Research Reagent Solutions for HSC Niche Modeling
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Biomaterial Scaffolds | Gelatin methacrylamide (GelMA), Hyaluronic acid (HA), Fibrin, Collagen I | Provide 3D structural support with tunable mechanical properties (0.3-35 kPa range) [7] |
| Cytokine Cocktails | SCF (100 ng/mL), TPO (100 ng/mL), FLT3-L (50-100 ng/mL), IL-6, SDF-1 | Maintain HSC self-renewal and prevent differentiation in culture [92] |
| Small Molecule Inhibitors/Activators | SR1 (StemRegenin 1), UM729, CHIR99021 (GSK-3 inhibitor) | Enhance HSC self-renewal and ex vivo expansion efficiency [92] |
| Stromal Cell Lines | OP9, MS-5, HK cells, Primary human MSCs | Provide critical niche cell contact and paracrine signaling support |
| Extracellular Matrix Components | Fibronectin, Laminin, Heparan sulfate proteoglycans, Osteopontin | Regulate HSC adhesion, retention, and quiescence through integrin signaling |
| Gene Editing Tools | CRISPR-Cas9, Lentiviral vectors (VSV-G pseudotyped), Transposon systems | Enable genetic modification of HSCs for mechanistic studies and gene therapy |
| Oxygen Control Systems | Tri-gas incubators (O₂, CO₂, N₂), Hypoxia chambers, Oxygen-sensing probes | Maintain physiological bone marrow hypoxia (1-4% O₂) for HSC maintenance |
A critical consideration in reagent selection is the impact on HSC biology. For instance, clinically used protocols with cytokines such as TPO, SCF and FLT3-L increase expression of the LMO2 protooncogene, thereby increasing possibilities for genotoxicity [92]. This highlights the importance of carefully evaluating even standard reagent choices for potential unintended consequences.
The field of HSC niche modeling stands at a transformative juncture, where interdisciplinary integration of biomaterials, microengineering, computational modeling, and stem cell biology is progressively enhancing the fidelity of in vitro systems to their native in vivo counterparts. Current advances in 3D biomimetic platforms have demonstrated promising capabilities for HSC expansion and disease modeling, yet challenges remain in standardizing these protocols and improving their predictive value for clinical outcomes.
Future directions will likely focus on several key areas: First, the development of more sophisticated multi-niche systems that integrate both vascular and endosteal components in spatially organized architectures. Second, the incorporation of immune cells and neural elements to better mimic the comprehensive bone marrow microenvironment. Third, the application of artificial intelligence and machine learning to analyze complex multi-omics data from these models and identify novel regulatory patterns. Finally, the establishment of standardized validation frameworks that systematically quantify the functional capacity of in vitro expanded HSCs will be essential for clinical translation.
As these technologies mature, they hold the potential to revolutionize not only basic research into hematopoietic biology but also clinical practice in stem cell transplantation and the treatment of hematological malignancies. By addressing the current technical hurdles in standardization and translation, the research community can accelerate the development of more effective therapies based on a fundamental understanding of the HSC niche.
Bench to Bedside: Validating Niche Function Through Heterochronic Studies, Lineage Tracing, and Clinical Correlations
The concept of the hematopoietic stem cell (HSC) niche, first proposed by Schofield in 1978, has fundamentally shaped our understanding of how specialized bone marrow microenvironments regulate stem cell self-renewal, differentiation, and aging [96]. This conceptual framework is particularly relevant when investigating hematopoiesis across the lifespan, as the functional decline of HSCs during aging represents a primary driver of immune deficiency, anemia, and hematological malignancies in the elderly [97] [20]. Within this paradigm, heterochronic transplantation—the transfer of aged HSCs into young recipients—serves as a powerful experimental tool to disentangle cell-intrinsic aging mechanisms from the influences of the extrinsic microenvironment [20].
The aged hematopoietic system is characterized by a pronounced myeloid bias, reduced lymphoid output, diminished reconstitution capacity, and an expanded but functionally impaired HSC pool [97] [20] [33]. While intrinsic regulators of HSC aging include genomic instability, metabolic deregulation, and epigenetic drift, the bone marrow microenvironment undergoes parallel degenerative changes that contribute significantly to hematopoietic decline [97] [98]. These changes include alterations in niche cellular composition, accumulation of bone marrow adipose tissue, and development of a pro-inflammatory milieu often termed "inflammaging" [20] [14]. This review synthesizes current evidence regarding the rejuvenating potential of young niches on aged HSCs, providing technical guidance for researchers investigating microenvironmental influences on hematopoietic aging.
Physiological Changes in the Aged Hematopoietic Niche
Cellular and Molecular Hallmarks of Niche Aging
The bone marrow niche undergoes multifaceted changes during aging that collectively impair its support capacity for HSCs. Key alterations include remodeling of mesenchymal stromal populations, vascular changes, and shifts in the inflammatory microenvironment [20] [98] [33]. Aged niches demonstrate a trending decrease in endosteal peri-arteriolar Sca-1+ mesenchymal stromal cells (MSC-S) and a significant reduction in both the frequency and fibroblast colony-forming unit (CFU-F) capacity of CD51+ osteoprogenitors [97]. Perhaps the most notable alteration is the accumulation of bone marrow adipocytes, which secrete adipokines like adiponectin that create a pro-inflammatory environment detrimental to normal hematopoietic function [14].
Table 1: Key Alterations in the Aged Hematopoietic Niche
| Niche Component | Young Niche Characteristics | Aged Niche Characteristics | Functional Consequences |
|---|---|---|---|
| Mesenchymal Stromal Cells | Maintained MSC-S population; active osteoprogenitors | Decreased MSC-S; reduced osteoprogenitor frequency and CFU-F capacity | Impaired HSC maintenance and support |
| Adipocytes | Limited bone marrow adipose tissue | Expanded adipose tissue; increased adiponectin secretion | Pro-inflammatory environment; altered DC function |
| Vascular Niche | Properly organized arteriolar and sinusoidal structures | Impaired vascular function; reduced Notch signaling | Compromised HSC quiescence and maintenance |
| Inflammatory Milieu | Balanced cytokine profile | Elevated pro-inflammatory factors (e.g., Ccl5, IL-6, IL-1β) | Myeloid bias; increased HSC proliferation |
| Megakaryocytes | Normal distribution and numbers | Expanded population | Potential alteration of HSC quiescence signals |
| Neural Components | Maintained sympathetic innervation | Diminished nerve density and function | Dysregulated HSC mobilization and maintenance |
Biophysical Changes in the Aged Niche
Beyond biochemical alterations, the aged niche exhibits significant biophysical changes that impact HSC function. The mechanical properties of cells and extracellular matrix (ECM)—including stiffness, viscoelasticity, and three-dimensional architecture—serve as fundamental physical regulators within the bone marrow hematopoietic microenvironment [7]. Matrix stiffness within the HSC niche is naturally heterogeneous, with the endosteal niche exhibiting a relatively rigid matrix (>35 kPa) and the vascular niche characterized by softer matrices (0.3-8 kPa) [7]. These mechanical properties dynamically influence HSC quiescence, differentiation, and migration through mechanotransduction mechanisms. Aging-associated changes in ECM composition and organization likely disrupt these mechanical cues, though the specific biophysical alterations in aged niches remain an active area of investigation.
Experimental Models for Assessing Niche Rejuvenation
Heterochronic Transplantation Approaches
Heterochronic transplantation represents the cornerstone experimental approach for investigating niche-mediated rejuvenation. The fundamental protocol involves isolating HSCs from aged donors (typically 20-24 month-old mice) and transplanting them into young, conditioned recipients (2-3 month-old mice), with subsequent analysis of hematopoietic reconstitution and HSC function [97] [20]. Proper controls include isochronic transplants (young-to-young and old-to-old) to establish baseline aging phenotypes.
Detailed Methodology:
- HSC Isolation: Purify Lin−/Sca-1+/c-Kit+/Flk2−/CD48−/CD150+ HSCs from aged C57BL/6-CD45.2 mice (24 months) and young controls (3 months) using fluorescence-activated cell sorting [97].
- Recipient Conditioning: Employ lethal irradiation (950 cGy) or non-genotoxic alternatives like CD45-saporin immunotoxin (3 mg/kg) for young syngeneic recipients [97] [99].
- Transplantation: Inject 250 purified HSCs via tail vein into conditioned recipients [97].
- Assessment: Track donor cell chimerism in peripheral blood over 16 weeks; analyze lineage distribution at 4 months post-transplantation; evaluate HSC function through secondary transplantation [97].
The gold standard for evaluating HSC regenerative function in this model includes assessing engraftment capacity, lymphoid-myeloid output balance, and secondary repopulating ability [97]. Transplantation into young mice without conditioning has also been explored using ex vivo expanded HSCs, demonstrating that niche availability rather than ablation can be sufficient for engraftment, though with limited lymphoid reconstitution [99].
Figure 1: Heterochronic Transplantation Experimental Workflow
Heterochronic Parabiosis and Plasma Transfer
Beyond transplantation, heterochronic parabiosis provides an alternative approach for investigating systemic influences on HSC aging. This surgical joining of circulatory systems between young and old animals allows assessment of how continuous exposure to young blood affects aged HSCs without the confounding effects of transplantation-associated stress [97] [100].
Detailed Methodology:
- Surgical Pairing: Surgically link 2-month-old C57BL/6-CD45.1 mice with 23-month-old C57BL/6-CD45.2 mice for heterochronic pairs, with isochronic pairs as controls [97].
- Duration: Maintain parabolic pairs for 4-5 weeks to allow substantial circulatory cross-talk [97].
- Separation and Analysis: Separate pairs and analyze HSC function and niche characteristics after varying recovery periods [97].
- Plasma Transfer: As a less invasive alternative, administer plasma from young donors to aged recipients (100-150μL injections 2-3 times weekly for 4 weeks) [100].
Notably, studies using these approaches have revealed that unlike other tissue stem cells, old HSCs show remarkable resistance to bloodborne systemic rejuvenation approaches, maintaining their cell-intrinsic aged state despite prolonged exposure to young blood or long-term residence in young niches after parabiont separation [97].
In Vitro Niche Modeling
Advanced in vitro systems enable reductionist approaches to dissect specific niche influences on HSC aging. These include three-dimensional biomimetic models, bone marrow-on-a-chip platforms, and conditioned media approaches that capture secretory profiles of young versus aged niches [7] [14].
Detailed Methodology for Conditioned Media Approach:
- Establish Niche Cultures: Isolate bone marrow cells from young (2-3 months) and aged (24 months) C57BL/6 mice [14].
- Long-term Culture: Resuspend BM cells in MyeloCult M5300 medium with hydrocortisone (1×10^−6 M) and seed at 1.1×10^6 cells/cm² [14].
- Maintenance: Culture in humidified CO2 incubator (33°C, 5% CO2) for 4 weeks, replacing half the medium weekly [14].
- Conditioned Media Collection: After 4 weeks, replace with serum-containing medium (RPMI-1640, 10% FBS, 1% Penicillin-Streptomycin) for 48 hours, then collect, centrifuge, and store conditioned media at -80°C [14].
- Functional Assays: Apply conditioned media to HSC differentiation or functional assays, such as dendritic cell development, tracking markers like MHC class II and CD86 [14].
This approach has demonstrated that the aged niche secretory profile promotes premature dendritic cell activation with elevated MHC class II expression and increased IL-6 secretion, indicating a heightened pro-inflammatory state [14].
Key Research Findings and Data Synthesis
Rejuvenation Capacity of Young Niches
The capacity of young niches to rejuvenate aged HSCs has yielded conflicting results across studies, with methodological differences likely explaining disparate findings. The table below synthesizes key quantitative findings from major studies investigating this question.
Table 2: Experimental Outcomes of Heterochronic Transplantation Studies
| Experimental Approach | Key Parameters Measured | Young-to-Young Control | Old-to-Old Control | Heterochronic (Old-to-Young) | Reference |
|---|---|---|---|---|---|
| Direct HSC Transplantation | Engraftment (%) | 85.2% ± 3.1 | 42.7% ± 5.8 | 45.1% ± 4.9 | [97] |
| Myeloid:Lymphoid Ratio | 1.5:1 | 4.8:1 | 4.6:1 | [97] | |
| Non-genotoxic Conditioning | Donor-derived HSC chimerism | 68.3% ± 7.2 | N/D | 12.4% ± 3.5 (aged recipients) | [99] |
| Ex Vivo Expanded HSCs | Multilineage engraftment without conditioning | 19.0% ± 2.1 (myeloid) | N/D | Limited lymphoid reconstitution | [99] |
| Heterochronic Parabiosis | HSC function post-separation | Maintained | Diminished | No improvement in old HSCs | [97] |
| In Vitro Niche Modeling | DC MHC II expression (MFI) | 1250 ± 180 | N/D | 2850 ± 320 (in aged conditioned media) | [14] |
The accumulated evidence suggests a complex picture regarding niche-mediated rejuvenation. While some studies indicate that young niches can partially reverse aging-associated phenotypes in HSCs [20], others demonstrate that old HSCs remain refractory to rejuvenation despite long-term exposure to young microenvironments [97]. This resistance appears to be maintained through cell-intrinsic mechanisms, including persistent replication stress marked by fibrillarin (FBL) and γH2AX double-positive nucleolar foci in old HSCs even after transplantation into young recipients [97].
Molecular Mechanisms of Niche-HSC Communication
The communication between HSCs and their niche involves multiple signaling pathways that become dysregulated with aging. Key pathways implicated in this cross-talk include Wnt signaling, TGF-β signaling, and inflammatory pathways such as mTOR activation [20] [100].
Figure 2: Signaling Pathways in Young vs. Aged HSC Niches
The aged niche exhibits elevated expression of chemokines like Ccl5 (RANTES), which activates mTOR signaling in HSCs and promotes myeloid bias [20]. Complement C1q, which accumulates with aging, acts as an agonist of canonical Wnt signaling by binding Frizzled receptors, leading to impaired tissue regeneration [100]. Additionally, adiponectin from expanded bone marrow adipose tissue in aged niches promotes pro-inflammatory dendritic cell activation [14]. In contrast, young niches provide balanced expression of stem cell factor (SCF) and CXCL12 that supports both myeloid and lymphoid differentiation while maintaining HSC quiescence [33].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Heterochronic Transplantation Studies
| Reagent/Category | Specific Examples | Application/Function | Technical Notes |
|---|---|---|---|
| Mouse Strains | C57BL/6-CD45.2, C57BL/6-CD45.1 | Congenic markers for tracking donor vs. recipient cells | CD45.1 recipients preferred for accepting CD45.2 donor cells |
| HSC Isolation Markers | Lin−, Sca-1+, c-Kit+, Flk2−, CD48−, CD150+ | Immunophenotypic identification and purification of HSCs | CD150 expression increases with HSC aging [97] |
| Conditioning Agents | CD45-saporin immunotoxin, Lethal irradiation (950 cGy) | Create niche space for donor HSC engraftment | Non-genotoxic conditioning preferred for aged models [99] |
| Culture Media | MyeloCult M5300 with hydrocortisone | Maintain niche cells for conditioned media production | Hydrocortisone final concentration 1×10^−6 M [14] |
| Flow Cytometry Antibodies | CD45.1, CD45.2, lineage panel (B220, CD3, etc.) | Chimerism analysis and lineage distribution | Essential for quantifying myeloid vs. lymphoid output |
| Cytokines/Antibodies | Recombinant SCF, TPO, CXCL12 | HSC expansion and functional assays | Young niches provide balanced cytokine expression [33] |
Discussion and Future Perspectives
The investigation of young niche effects on aged HSCs reveals the remarkable complexity of hematopoietic aging. While the weight of evidence suggests that old HSCs maintain strong cell-intrinsic aging programs resistant to microenvironmental rejuvenation [97], certain interventions demonstrate that functional improvement is possible under specific conditions. The successful engraftment of ex vivo expanded young HSCs in aged recipients using non-genotoxic conditioning represents a promising therapeutic avenue [99], though the persistence of aged HSC resistance in heterochronic transplantation remains a significant challenge.
Future research directions should focus on several key areas. First, the development of more sophisticated in vitro niche models that better recapitulate the three-dimensional architecture and multicellular composition of native bone marrow will enable more precise dissection of specific niche influences [7]. Second, exploring combinatorial approaches that simultaneously target both intrinsic HSC aging mechanisms and extrinsic niche factors may yield synergistic benefits. Third, translational efforts should prioritize non-genotoxic conditioning regimens that are better tolerated by aged individuals, potentially making HSC transplantation a viable prophylactic intervention for age-related hematological disorders [99].
The broader implication of this research extends beyond hematopoiesis to the fundamental biology of aging. The demonstrated resistance of HSCs to rejuvenating signals that effectively restore function in other tissue stem cells suggests unique mechanisms safeguarding the blood system from extrinsic perturbations [97]. Understanding these protective mechanisms may reveal novel targets for preventing age-related hematopoietic decline while offering insights into why the blood system is particularly vulnerable to certain aging-associated pathologies like clonal hematopoiesis and myeloid malignancies.
As we approach the 50th anniversary of Schofield's niche hypothesis, establishing consensus definitions and standardized methodologies for niche research will be crucial for advancing the field [96] [101]. The heterochronic transplantation paradigm continues to provide invaluable insights into the dynamic interplay between stem cells and their microenvironments throughout the lifespan, moving us closer to therapeutic interventions that can maintain robust hematopoiesis into advanced age.
Emergency hematopoiesis is a critical physiological response to acute insults such as myocardial infarction (MI), severe infection, or other systemic stressors. This process involves the rapid activation of hematopoietic stem and progenitor cells (HSPCs) within the bone marrow (BM) microenvironment to increase the production of immune cells, particularly those of the myeloid lineage [102] [103]. Following MI, the heightened demand for leukocytes to participate in inflammatory responses and tissue repair at the injury site triggers a cascade of signaling events that activate normally quiescent HSPCs [102]. Understanding the precise contributions of specific HSC subsets and their regulatory mechanisms during this process requires sophisticated lineage tracing models that can map cellular fate decisions with high resolution. This technical guide explores the current methodologies, key findings, and experimental protocols defining how lineage tracing models elucidate HSC dynamics in emergency hematopoiesis, with particular emphasis on post-MI contexts.
Table 1: Key Hematopoietic Stem and Progenitor Cell (HSPC) Subsets in Emergency Hematopoiesis
| Cell Population | Phenotypic Markers (Mouse) | Proliferative Status | Myeloid Bias | Functional Role in Emergency Hematopoiesis |
|---|---|---|---|---|
| CCR2+ HSPC | Lin⁻ Sca-1⁺ c-Kit⁺ (LSK), CD150⁺ CD48⁻, CCR2⁺ | High proliferation post-MI (≥40% BrdU⁺) [102] | Yes [102] | Drives emergency myelopoiesis; dominates migratory HSPC pool [102] |
| CCR2- HSC | Lin⁻ Sca-1⁺ c-Kit⁺ (LSK), CD150⁺ CD48⁻, CCR2⁻ | Predominantly quiescent post-MI [102] | No [102] | Maintains long-term, multilineage reconstitution capacity [102] |
| P2Y12+ LSK | Lin⁻ Sca-1⁺ c-Kit⁺, P2Y12 receptor expression | ADP-mediated Akt phosphorylation and cell cycle progression [103] | Promotes myeloid output [103] | Fuels post-ischemic inflammation via purinergic signaling [103] |
The Bone Marrow Niche: Foundation for HSC Regulation
The bone marrow niche represents a sophisticated microenvironment that provides both structural and biochemical support to regulate HSC function, balancing quiescence, self-renewal, and differentiation [47] [8]. This specialized compartment consists of multiple cellular components, including mesenchymal stromal cells (MSCs), endothelial cells, osteoblasts, macrophages, and neural cells, which interact with HSCs through direct contact and secreted factors [104] [47] [8].
The BM contains at least two distinctive HSC-supportive niches: the endosteal niche, primarily associated with osteoblasts and supporting HSC quiescence, and the vascular niche, composed of sinusoidal and arteriolar endothelial cells that promote proliferation and differentiation [47]. These niches employ key signaling molecules such as CXCL12, stem cell factor (SCF), thrombopoietin (TPO), and transforming growth factor-beta (TGF-β) to maintain HSC homeostasis [47] [8] [5]. During emergency hematopoiesis, this carefully balanced microenvironment undergoes significant modification, with altered cytokine expression and cellular interactions that promote HSPC activation and myeloid-biased differentiation [102] [103].
Lineage Tracing Technologies: Mapping Hematopoietic Fate Decisions
Lineage tracing encompasses a suite of techniques designed to establish hierarchical relationships between cells and track their progeny over time. Recent technological advancements have dramatically improved our ability to resolve hematopoietic lineages at single-cell resolution.
Historical Development and Traditional Approaches
The foundation of modern lineage tracing was established with site-specific recombinase (SSR) systems, particularly Cre-loxP, which allows for permanent genetic labeling of specific cell populations and their descendants [105]. When combined with inducible systems (e.g., CreERT2), this technology enables temporal control over labeling initiation using agents like tamoxifen [105].
Multicolor lineage tracing approaches, such as Brainbow and R26R-Confetti, represent significant advancements by employing stochastic recombination events to generate multiple fluorescent hues within a cell population [105] [106]. This allows simultaneous tracking of numerous clones, providing insights into clonal dynamics and cellular interactions within complex tissues [105].
Single-Cell Lineage Tracing (SCLT) Platforms
The integration of single-cell sequencing technologies with lineage tracing has created powerful tools for dissecting hematopoietic heterogeneity:
- Integration Barcodes: Utilizing retroviral vectors to introduce unique, heritable DNA sequences into HSPCs, enabling long-term tracking of clonal outputs and relationships [106].
- CRISPR Barcoding: Employing CRISPR/Cas9 to induce cumulative insertions and deletions (InDels) in synthetic genomic loci, which serve as genetic recorders of cell division history and lineage relationships [106].
- Polylox Barcodes: An artificial DNA recombination system based on Cre-loxP that enables in vivo barcoding through combinatorial recombination events [106].
- Natural Barcodes: Leveraging naturally accumulating somatic mutations as endogenous markers for lineage reconstruction, applicable to human studies without genetic modification [106].
Table 2: Comparison of Single-Cell Lineage Tracing Technologies
| Method | Mechanism | Key Advantages | Limitations |
|---|---|---|---|
| Integration Barcodes | Retroviral vector insertion of random sequence tags [106] | High information quantity and accuracy; tracks thousands of clones simultaneously [106] | Limited to proliferating cells; potential for viral silencing [106] |
| CRISPR Barcodes | CRISPR/Cas9-induced InDels and mutations in synthetic genomic loci [106] | High mutation rate records extensive mitotic history; detailed phylogenetic trees [106] | Not suitable for human primary cells; complex data analysis [106] |
| Polylox Barcodes | Cre-loxP-mediated recombination of artificial DNA locus [106] | High specificity; labels single progenitor cells in vivo [106] | Not suitable for human primary cells; requires Cre expression [106] |
| Natural Barcodes | Endogenous somatic mutations accumulated during development and aging [106] | Applicable to human primary cells; non-invasive [106] | Immature sequencing methods; limited by natural mutation rate [106] |
Emergency Hematopoiesis Post-Myocardial Infarction: Key Insights from Lineage Tracing
CCR2+ HSPC Subset Activation
Research utilizing sophisticated fate-mapping models has revealed that MI triggers a selective activation of specific HSPC subsets rather than uniform activation of all HSCs. A CCR2+ CD150+ CD48- LSK population has been identified as the primary driver of emergency hematopoiesis post-MI [102]. These cells demonstrate significantly higher proliferation rates compared to their CCR2- counterparts, with >40% incorporating BrdU within 48 hours of coronary ligation [102].
This CCR2+ HSPC subset exhibits distinct functional properties, including myeloid differentiation bias and reduced self-renewal capacity upon secondary transplantation [102]. Intravital microscopy studies have revealed spatial organization correlates with functional heterogeneity: while CCR2- HSCs predominantly reside near the endosteum, CCR2+ HSPCs localize more distantly from bone surfaces, reflecting their activated state [102].
Purinergic Signaling in HSPC Activation
Beyond chemokine receptors, purinergic signaling has emerged as a critical regulator of emergency hematopoiesis. The P2Y12 receptor, traditionally associated with platelet activation, is functionally expressed on LSK cells and responds to ADP released following tissue injury [103]. P2Y12 activation promotes Akt phosphorylation and cell cycle progression in HSPCs, thereby fueling emergency hematopoiesis [103].
Pharmacological inhibition or genetic ablation of P2Y12 signaling reduces HSPC proliferation and myeloid output post-MI, resulting in attenuated inflammatory responses and improved cardiac remodeling [103]. This highlights the potential for non-canonical effects of P2Y12 antagonists beyond platelet inhibition.
Systemic Regulation of HSC Numbers
Recent research challenges the classical niche model by demonstrating that HSC numbers are regulated at both systemic and local levels, independent of niche availability alone [5]. Thrombopoietin (TPO) has been identified as a pivotal systemic regulator determining total HSC numbers in the body [5]. This discovery has significant implications for understanding how emergency hematopoiesis is controlled globally during systemic stressors like MI.
Experimental Protocols for Investigating HSC in Emergency Hematopoiesis
Protocol 1: Myocardial Infarction Model and HSPC Analysis
Objective: To assess HSPC activation and lineage contributions following ischemic cardiac injury.
Materials:
- C57BL/6 mice (8-13 weeks old)
- Anesthetics: ketamine (100 mg/kg) and xylazine (10 mg/kg)
- Analgesia: buprenorphine (0.1 mg/kg)
- Surgical instruments for thoracotomy
- 8-0 prolene suture for coronary artery ligation
Methodology:
- Induce anesthesia and administer preoperative analgesia.
- Perform endotracheal intubation and maintain ventilation with isoflurane (0.5-2%).
- Execute left lateral thoracotomy between the 3rd and 4th rib.
- Identify the left anterior descending (LAD) coronary artery and perform permanent ligation in its proximal middle third.
- Close thoracic walls and skin with 5-0 prolene suture.
- At designated timepoints (e.g., 48 hours post-MI), administer BrdU or analyze cell cycle status.
- Harvest femurs, tibiae, and pelvis for BM analysis.
- Generate single-cell suspensions by flushing bones and filtering through 40μm strainers.
- Perform flow cytometry staining for HSC populations (Lin⁻ Sca-1⁺ c-Kit⁺, CD150⁺ CD48⁻) and CCR2 expression.
- Assess proliferation via BrdU incorporation or cell cycle analysis [102].
Protocol 2: P2Y12 Signaling in Emergency Hematopoiesis
Objective: To evaluate ADP-P2Y12 axis contribution to HSPC activation post-MI.
Materials:
- Global P2Y12-knockout mice or platelet-specific P2Y12-deficient mice
- P2Y12 inhibitor (e.g., prasugrel, 5 mg/kg body weight)
- ADP for in vitro stimulation
- Antibodies for phospho-Akt flow cytometry
Methodology:
- Treat mice with prasugrel or vehicle one day prior to MI induction.
- Induce MI as described in Protocol 1.
- Continue daily prasugrel administration post-MI.
- Isolate LSK cells from BM for in vitro ADP stimulation studies.
- Assess Akt phosphorylation via flow cytometry.
- Analyze cell cycle progression in LSK populations.
- Quantify downstream myeloid progeny in blood and infarcted myocardium.
- Evaluate cardiac function and remodeling by echocardiography [103].
Signaling Pathways in Emergency Hematopoiesis
Figure 1: Signaling Pathways in Post-MI Emergency Hematopoiesis. This diagram illustrates the key molecular and cellular events connecting myocardial infarction to hematopoietic stem cell activation and subsequent systemic effects.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Emergency Hematopoiesis Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Genetic Mouse Models | CCR2-RFP reporters [102], P2Y12 global KO [103], Nestin-GFP [5], Cdh5-CreER [5] | Enable cell-type-specific labeling, tracking, and functional manipulation of HSCs and niche components |
| Lineage Tracing Systems | Cre-loxP [105] [106], Dre-rox [105], R26R-Confetti [105], Polylox barcodes [106] | Provide permanent genetic labeling of HSCs and their progeny for fate mapping |
| Flow Cytometry Antibodies | Anti-CD150, anti-CD48, anti-Sca-1, anti-c-Kit, anti-CCR2, anti-CD45 isoforms [102] [5] | Identification and purification of specific HSC subsets and differentiated progeny |
| Pharmacological Inhibitors | Prasugrel (P2Y12 antagonist) [103], G-CSF [5] | Modulate specific signaling pathways involved in HSC activation and mobilization |
| Cell Tracking Agents | BrdU, EdU [105] [102], fluorescent membrane dyes (e.g., CM-DiI) [102] | Assess proliferation history and track cell localization/migration |
| Single-Cell Technologies | 10X Genomics, Smart-seq2 [106], CRISPR barcoding systems [106] | Enable high-resolution analysis of clonal dynamics and heterogeneity |
Future Directions and Therapeutic Implications
Advanced lineage tracing models continue to reveal the remarkable heterogeneity within the HSC pool and their differential contributions to emergency hematopoiesis. The identification of distinct HSPC subsets with specialized roles in stress responses opens new avenues for therapeutic intervention. Targeting specific subpopulations, such as CCR2+ HSPCs or P2Y12-mediated signaling, may allow for more precise modulation of emergency hematopoiesis without compromising steady-state hematopoiesis [102] [103].
Future research directions include developing more sophisticated multicolor and multi-recombinase systems for simultaneous tracking of multiple lineages, improving CRISPR-based recording technologies for longer-term lineage tracing, and integrating single-cell transcriptomic data with lineage information to correlate fate decisions with molecular signatures [105] [106]. Additionally, understanding how niche-derived extracellular vesicles and systemic factors like TPO coordinate local and global regulation of HSC numbers during stress conditions will be crucial for developing comprehensive models of emergency hematopoiesis [107] [5].
As these technologies advance, they will provide increasingly refined insights into HSC biology, potentially revolutionizing therapeutic approaches for conditions ranging from myocardial infarction to hematologic malignancies where dysregulated emergency hematopoiesis contributes to disease pathology.
The bone marrow (BM) microenvironment, or hematopoietic stem cell (HSC) niche, is a complex, spatially organized tissue where precise cellular interactions govern blood production and disease pathogenesis. For decades, understanding this architecture in human patient samples remained challenging due to technological limitations. The advent of single-cell RNA sequencing (scRNA-seq) revolutionized our ability to dissect cellular heterogeneity but sacrificed crucial spatial context. Today, spatial transcriptomics (ST) has emerged as a transformative technology that preserves this geographical information, enabling researchers to map gene expression patterns within the intact tissue architecture. When integrated, these technologies provide an unprecedented view of the BM niche's spatial and functional organization, offering new insights into both normal hematopoiesis and disease states such as acute myeloid leukemia (AML) and multiple myeloma (MM) [108] [109].
This technical guide explores the synergy of scRNA-seq and ST for mapping the human BM niche. We detail experimental and computational methodologies, showcase applications in malignant hematopoiesis, and provide a practical toolkit for implementing these approaches in translational research.
Technology Landscape: From Single-Cell Dissociation to Spatial Mapping
Single-Cell RNA Sequencing (scRNA-seq)
scRNA-seq analyzes gene expression profiles of individual cells from heterogeneous populations. By isolating single cells—typically through encapsulation or flow cytometry—followed by RNA amplification and sequencing, researchers can identify and characterize different cell types, states, and subpopulations with exceptional resolution [110]. A key advantage over bulk RNA sequencing is the ability to detect rare cell subtypes and gene expression variations that would otherwise be averaged out [111] [110]. The standard workflow involves tissue dissociation, single-cell capture, RNA reverse transcription, cDNA amplification, and library preparation for sequencing [111].
Table 1: Comparison of Bulk RNA-seq and Single-Cell RNA-seq
| Feature | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Average gene expression across thousands of cells | Gene expression profiles of individual cells |
| Spatial Context | Lost during tissue dissociation | Lost during tissue dissociation |
| Cellular Heterogeneity | Masked | Revealed |
| Key Application | Identifying population-level expression changes | Discovering rare cell types, reconstructing developmental trajectories |
Spatial Transcriptomics (ST) Platforms
Spatial transcriptomics encompasses a suite of technologies that retain the spatial localization of RNA transcripts within tissue sections. These methods can be broadly classified into two categories: sequencing-based approaches that decode spatial barcodes, and imaging-based methods that visualize in situ mRNA [112]. These technologies have been systematically benchmarked using reference tissues, revealing significant variability in parameters like molecular diffusion, capture efficiency, and effective resolution across different platforms [113].
Table 2: Key Spatial Transcriptomics Technologies and Their Performance
| Technology | Methodology | Resolution | Key Strengths | Challenges in BM |
|---|---|---|---|---|
| 10x Visium [109] | Sequencing-based (spatial barcoding) | 55 μm (captures 3-10 cells/spot) | Whole transcriptome, compatible with FFPE | Low single-cell resolution, spot-level data requires deconvolution |
| Slide-seqV2 [113] | Bead-based sequencing | ~10 μm (near single-cell) | Higher resolution than Visium | Lower capture efficiency, requires advanced computational analysis |
| Stereo-seq [113] | Polony-based sequencing | <10 μm (subcellular) | High resolution and massive capture area | Extremely high sequencing depth required |
| MERFISH [112] | Imaging-based (in situ hybridization) | Subcellular | Single-cell resolution, high accuracy | Targeted approach (limited gene panel) |
| DBiT-seq [113] | Microfluidics-based | 20-50 μm | Combines protein and RNA detection | Limited capture area |
Overcoming Bone-Specific Technical Challenges
Working with human BM biopsies presents unique technical hurdles that must be addressed for successful spatial transcriptomic analysis.
Specialized Tissue Processing Protocol
BM core biopsies require careful processing to preserve both histology and RNA integrity while dealing with tissue mineralization [109].
- Decalcification: Following fixation, BM biopsies must undergo decalcification to remove mineralized bone matrix. This step is crucial for sectioning but can compromise RNA integrity. Use gentle, RNA-friendly decalcifying agents (e.g., EDTA-based solutions) over shorter durations [109].
- Formalin-Fixed Paraffin-Embedding (FFPE): While FFPE preservation is standard in clinical pathology, it introduces RNA cross-linking and fragmentation. Protocols using the Visium Spatial Gene Expression for FFPE platform have been successfully applied to both mouse and human BM samples, including from Multiple Myeloma patients [109].
- Sectioning and Placement: For technologies like Visium, tissue sections (typically 5-10 μm thick) are placed onto patterned, spatially barcoded oligonucleotide arrays. Precise orientation and sectioning through the trabecular bone region are essential, as the cortical region often yields minimal counts [109].
Addressing Data Analysis Challenges
BM spatial data is characterized by low cellularity and high adipocyte content, leading to potential gene dropouts and technical noise [109]. A custom, data-driven analytical framework is recommended, which includes:
- Stringent Quality Control: Applying custom thresholds for minimum counts/features per spot to exclude low-quality data.
- Cell Type Deconvolution: Using matched scRNA-seq data as a reference to estimate the proportion of different cell types within each spatial spot. This is critical for interpreting spot-level data from platforms like Visium [109].
- Spatial Neighborhood Analysis: Defining regions of interest (e.g., tumor-rich areas, endosteal regions) based on cell type composition rather than pre-defined anatomical landmarks.
Integrated Experimental Workflow for Niche Mapping
The power of modern niche analysis lies in combining scRNA-seq and ST.
Integrated scRNA-seq and ST Workflow
Protocol: Multi-Modal Analysis of Patient BM
Step 1: Parallel Sample Processing Split a single BM core biopsy into two portions. One portion is dissociated into a single-cell suspension for scRNA-seq. The other portion is fixed, decalcified, and embedded in FFPE or OCT compound for spatial transcriptomics [109].
Step 2: Library Preparation and Sequencing
- For scRNA-seq: Use a platform like 10x Genomics Chromium to generate barcoded libraries from the single-cell suspension.
- For ST: Mount tissue sections on the chosen spatial platform (e.g., Visium slide). Perform H&E staining and imaging, followed on-slide cDNA synthesis and library preparation [109].
Step 3: Computational Data Integration
- Alignment and Clustering: Process scRNA-seq data to identify distinct cell populations and generate a reference transcriptome profile for each cell type.
- Spatial Data Mapping: Use the scRNA-seq data as a reference to deconvolute the spot-based spatial data, estimating the cellular composition of each location [109].
- Spatially Resolved Analysis: Identify spatially variable genes and analyze cell-cell communication patterns within their geographical context.
Application: Illuminating the Niche in Hematologic Malignancies
Acute Myeloid Leukemia (AML) and Immunotherapy
A seminal study used a multi-omic approach to investigate the BM niche in patients with refractory/relapsed AML treated with pembrolizumab and decitabine. The researchers integrated scRNA-seq data with single-cell-resolution spatial transcriptomic data from the same sample [108]. Key findings included:
- Niche Remodeling: Post-immunotherapy, multiple immune cell types showed global or local enrichment near leukemia cells.
- Accurate Cell-Cell Distance Mapping: By quantifying distances between cell edges rather than centroids, the study achieved a more accurate analysis of the tumor microenvironment [108].
- Signaling Alterations: Ligand-receptor analysis suggested a potential increase in TWEAK signaling between leukemia and immune cells after treatment, revealing a possible resistance mechanism [108].
Multiple Myeloma (MM) Microenvironment
Spatial transcriptomics of MM patient BM has revealed profound remodeling of the niche. Using Visium on FFPE samples, researchers characterized:
- T-cell Exhaustion Gradients: A spatial gradient was observed where T cells transitioned from an effector to an exhausted phenotype as their distance from MM plasma cells increased [109].
- Spatially Distinct Signaling: Malignant plasma cell-rich regions showed reduced signatures of NETosis and IL-17 signaling, indicating localized immunosuppression [109].
- Therapy Resistance: Interactions between MM cells and mesenchymal stromal cells (MSCs), including mitochondrial transfer via tunneling nanotubes, have been spatially mapped, revealing a mechanism of drug resistance [114].
Clonal Hematopoiesis and Inflammatory Remodeling
Spatial analysis of BM from patients with clonal hematopoiesis (CHIP) and myelodysplastic syndromes (MDS) has uncovered a role for chronic inflammation in niche dysregulation. A recent study found:
- Inflammatory Stromal Cells: A population of inflammatory MSCs (iMSCs) gradually replaces normal supportive MSCs. These iMSCs produce interferon-induced cytokines, creating a feed-forward loop with interferon-responsive T cells [75].
- Loss of Protective Signals: MDS stem cells failed to trigger CXCL12 production in stromal cells, a key signal for normal hematopoiesis, explaining marrow failure [75].
Inflammatory Niche Remodeling in Myeloid Malignancy
Successfully mapping the BM niche requires both wet-lab reagents and sophisticated computational tools.
Table 3: Research Reagent Solutions for Niche Mapping
| Item | Function | Example/Note |
|---|---|---|
| Gentle MACS Dissociator | Mechanical tissue dissociation for viable single-cell suspension | Preserves cell viability for scRNA-seq |
| RNA-Friendly Decalcification Solution | Removes bone mineral while preserving RNA integrity | EDTA-based, avoid strong acids |
| Visium Spatial Tissue Optimization Slide | Pre-test RNA quality of FFPE sections | Critical for sample qualification |
| Visium Spatial for FFPE Reagent Kit | Whole transcriptome analysis from FFPE tissue | Includes probe design for fragmented RNA |
| Antibody-Derived Tags (ADT) | Surface protein quantification alongside scRNA-seq | CITE-seq to resolve immune cell states |
| Cell Hashtag Oligonucleotides | Multiplexing samples in one scRNA-seq run | Reduces batch effects and costs |
Computational Tools for Data Analysis
- Deconvolution Methods: Tools like Tangram [112] and Cell2location are used to map cell types from scRNA-seq onto spatial data.
- Spatial Analysis Pipelines: Seurat [112] and Giotto provide frameworks for identifying spatially variable genes and analyzing cellular neighborhoods.
- Cell-Cell Communication: CellChat and NicheNet can infer ligand-receptor interactions within the spatial context.
- Custom Scripting: As shown in the MM study [109], custom R/Python scripts are often needed for thresholding, region definition, and analysis of low-cellularity BM data.
The integration of single-cell and spatial transcriptomics has fundamentally advanced our understanding of the human bone marrow niche. These technologies have moved us from a simplistic model of HSC localization to a nuanced appreciation of dynamic, disease-specific microenvironments that actively shape therapeutic responses. The future of niche mapping lies in increasing resolution through platforms like Stereo-seq, combining transcriptomic with proteomic and epigenetic data, and implementing longitudinal studies to track niche evolution during treatment. As these tools become more accessible, they will undoubtedly uncover new therapeutic targets that disrupt pathogenic niches while preserving normal hematopoiesis, ultimately improving outcomes for patients with hematologic malignancies.
The bone marrow hematopoietic stem cell (HSC) niche undergoes profound functional and structural alterations with aging that significantly impact immune cell output and function. This progressive remodeling of the bone marrow microenvironment contributes to immunosenescence and inflammaging, characterized by diminished adaptive immune responses and chronic low-grade inflammation. Recent research demonstrates that the aged HSC niche directly influences the development and functionality of immune cells, particularly dendritic cells (DCs), through altered secretory profiles, skewed differentiation patterns, and modified cell-cell interactions. This review synthesizes current understanding of how young versus aged HSC niches differentially regulate immune cell output, with emphasis on DC function, and discusses implications for therapeutic interventions targeting niche-driven immune dysfunction in aging.
The HSC niche constitutes a specialized bone marrow microenvironment that regulates hematopoietic stem cell maintenance, self-renewal, and differentiation through complex cellular interactions and molecular signaling. This microenvironment includes vascular components (arteriolar and sinusoidal endothelial cells), neural components (sympathetic nerves, non-myelinating Schwann cells), stromal components (mesenchymal stromal cells, osteoblasts, adipocytes), and hematopoietic components (HSCs and their progeny) [14]. The coordinated function of these elements ensures balanced production of all blood cell lineages, including immune cells.
With advancing age, the HSC niche undergoes significant changes that disrupt its normal regulatory functions. One of the most notable alterations is the expansion of bone marrow adipose tissue and increased adipocyte accumulation, which has been linked to impaired hematopoiesis and altered immune cell production [14] [20]. Additionally, aging induces changes in the spatial organization of niche components, modifications in secretory profiles, and alterations in cell-cell communication. These age-related transformations create a microenvironment that differentially supports immune cell development compared to young niches, ultimately contributing to the decline in immune function observed in older individuals.
Age-Related Transformations of the HSC Niche
Structural and Cellular Changes
The bone marrow microenvironment experiences multiple structural and cellular alterations during the aging process that collectively impact its ability to support normal hematopoiesis:
Adipocyte Expansion: A hallmark of the aging bone marrow is the significant increase in adipocyte number and size. This marrow adipose expansion occurs at the expense of functional hematopoietic tissue and has been correlated with impaired hematopoietic support capacity [14] [20]. Adipocytes in aged marrow exhibit altered secretory profiles, releasing different levels of adipokines such as adiponectin, which can modulate immune cell function.
Megakaryocyte Expansion: Multiple studies have documented an increase in megakaryocytes and megakaryocyte progenitors in aged bone marrow [20]. These cells, which normally regulate HSC quiescence through factors like CXCL4, may contribute to the functional decline of HSCs when their numbers and spatial relationships are altered.
Stromal Cell Dysfunction: Mesenchymal stromal cells in aged niches show reduced capacity to support lymphopoiesis and exhibit altered expression of key hematopoietic factors including CXCL12 and stem cell factor (SCF) [20] [5]. Additionally, increased stromal stiffness has been observed in aged bone marrow, rising from approximately 3 kPa in young adulthood to 8 kPa with aging, which affects HSC function through mechanosensitive signaling pathways [25].
Vascular Alterations: Aging is associated with changes in the bone marrow vasculature, though research findings regarding specific numerical changes have been inconsistent. What remains clear is that functional alterations in endothelial cells impair their ability to support HSC maintenance and proper differentiation [20].
Molecular and Secretory Profile Alterations
The aged HSC niche exhibits a distinctly different molecular profile compared to its young counterpart, characterized by:
Pro-inflammatory Shift: Analysis of conditioned media from in vitro HSC niche models revealed that aged niches promote a pro-inflammatory state, with increased presence of factors like adiponectin and other inflammatory mediators [14]. This inflammatory microenvironment contributes to "inflammaging" - the chronic, low-grade inflammation characteristic of aging.
Altered Cytokine and Chemokine Expression: Aged niches show dysregulated expression of key hematopoietic cytokines. For instance, elevated levels of Ccl5 (RANTES) in the aged microenvironment have been shown to induce myeloid bias in young HSCs, mirroring the differentiation pattern observed in aged hematopoiesis [20]. Conversely, some supportive factors may be decreased in aged niches.
Senescence-Associated Secretory Phenotype (SASP): Senescent stromal and hematopoietic cells in aged niches secrete a collection of pro-inflammatory cytokines, growth factors, and proteases known as SASP. This includes elevated levels of IL-6, IL-8, and TNF-α, which further perpetuate inflammation and tissue dysfunction [115].
Table 1: Key Molecular Alterations in the Aged HSC Niche
| Molecular Factor | Change with Aging | Functional Consequences |
|---|---|---|
| Adiponectin | Increased [14] | Promotes premature DC activation; modulates inflammation |
| Ccl5 (RANTES) | Increased [20] | Induces myeloid bias in HSCs via mTOR activation |
| IL-6 | Increased [14] [115] | Promotes chronic inflammation; alters DC function |
| CXCL12 | Context-dependent alterations [20] [5] | Affects HSC retention and lymphoid differentiation |
| SASP Factors | Increased [115] | Creates pro-inflammatory microenvironment |
Impact on Hematopoietic Stem Cell Function and Immune Output
HSC Functional Decline and Lineage Skewing
Aged HSCs residing in aged niches exhibit several functional deficits that directly impact immune cell output:
Myeloid Differentiation Bias: A hallmark of hematopoietic aging is the skewing of differentiation potential toward myeloid lineages at the expense of lymphoid output. This myeloid bias results in increased production of granulocytes and monocytes and decreased generation of lymphocytes, particularly B cells [20] [25]. Single-cell transplantation studies have revealed a reduced proportion of lympho-biased stem cells and an expansion of myeloid-restricted progenitor cells in aged HSCs [25]. This bias has significant implications for immune competence, as it reduces the diversity of the adaptive immune repertoire.
Impaired Self-Renewal: Aged HSCs demonstrate reduced reconstructive capacity in transplantation assays, indicating diminished self-renewal potential [25]. This functional decline arises from both cell-intrinsic mechanisms and extrinsic factors from the aged microenvironment.
Altered Spatial Distribution: Aged HSCs have been observed to lodge further from the endosteum after homing and exhibit changes in their spatial relationship with niche cells like megakaryocytes, potentially affecting the signaling they receive [20].
The molecular mechanisms driving these changes include epigenetic alterations such as elevated HDAC3 activity leading to deacetylation at the H4K77 site, which directly inhibits expression of key lymphoid differentiation genes like EBF1 and PAX5 [25]. Additionally, metabolic shifts toward fatty acid oxidation and defects in mitochondrial quality control further promote myeloid differentiation.
Systemic Regulation of HSC Numbers
Recent research challenges the classical view that HSC numbers are determined primarily by niche availability. Studies using femur transplantation systems demonstrate that adding new niches does not increase total HSC numbers in the body, suggesting the presence of systemic regulators that maintain HSC numbers within a fixed range [5]. Thrombopoietin has been identified as a pivotal factor in determining the total number of HSCs in the body, even when niche availability is increased [5]. This insight refines our understanding of how HSC numbers are regulated during aging and suggests that functional quality rather than quantity may be the primary determinant of immune output efficacy.
Differential Impact on Dendritic Cell Development and Function
Experimental Models for Assessing Niche Effects on DCs
To directly compare how young versus aged HSC niches affect dendritic cell biology, researchers have developed sophisticated in vitro models that recapitulate key aspects of these microenvironments:
Conditioned Media Approach: Bone marrow cells from young (2-3 months) and aged (24 months) mice are cultured in long-term culture medium for 4 weeks to establish in vitro HSC niche models [14]. Conditioned media is collected from these cultures and used in BM-derived DC (BMDC) differentiation and maturation protocols, allowing assessment of niche-specific soluble factors on DC development.
Biomimetic 3D Culture Systems: Recent advances include development of three-dimensional biomimetic models using 3D printing, organoids, and bone marrow-on-a-chip platforms to more accurately replicate the native bone marrow architecture [28]. These systems enable study of both soluble factors and cell-cell contacts in DC development.
Table 2: Experimental Approaches for Studying Niche-DC Interactions
| Method | Key Components | Applications | Advantages |
|---|---|---|---|
| Conditioned Media | Soluble factors from young/aged niche cultures [14] | BMDC differentiation and maturation studies | Isolates effects of secreted factors; technically accessible |
| 3D Biomimetic Niches | Biomaterials, stromal cells, ECM components [28] | Study of complex niche-DC interactions | Presents structural and cellular complexity; more physiologically relevant |
| Bone Marrow Transplantation | Young HSCs in aged recipients and vice versa [20] | In vivo analysis of niche effects on hematopoiesis | Preserves full physiological context; complex interpretation |
Functional Consequences for Dendritic Cells
Research using these models has revealed significant differences in how young versus aged HSC niches shape DC biology:
Premature Activation: BMDCs differentiated in aged niche-conditioned media exhibit characteristics of premature activation, including elevated MHC class II expression and enhanced allostimulatory capacity at their immature stage [14]. This suggests that aged niche factors drive DCs toward an activated state even in the absence of overt inflammatory signals.
Altered Response to Maturation Signals: Upon LPS stimulation (used to induce DC maturation), BMDCs from the aged niche environment showed significantly increased CD86 expression compared to those from young niches [14]. However, despite this heightened surface marker expression, these cells did not demonstrate superior allostimulatory capacity, indicating a disconnect between phenotypic maturation and functional efficacy in DCs from aged niches.
Pro-inflammatory Polarization: Analysis of cytokine profiles revealed that BMDCs cultured in aged niche-conditioned media secreted significantly higher levels of IL-6, indicating a heightened pro-inflammatory activation state [14]. This pro-inflammatory skewing may contribute to the chronic inflammatory state observed in aged individuals and could potentially drive inappropriate immune activation.
Metabolic and Functional Alterations: While direct evidence from the provided studies is limited, broader aging research suggests that DCs from aged environments exhibit metabolic shifts and functional impairments in antigen processing and presentation, further compromising adaptive immune responses [115].
Figure 1: Mechanisms of DC Dysregulation in Aged HSC Niches. The aged niche environment promotes DC dysfunction through multiple interconnected pathways.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Niche-Immune Cell Interactions
| Reagent/Cell System | Specifications | Research Application | Key Functions |
|---|---|---|---|
| MyeloCult M5300 | Complete long-term culture medium with hydrocortisone [14] | In vitro HSC niche modeling | Supports maintenance of hematopoietic cells and stromal components |
| C57BL/6JRccHsd Mice | Young (2-3 months) vs. aged (24 months) females [14] | Aged hematopoiesis studies | Standardized model for aging research; well-characterized immune system |
| BALB/c Mice | 6-7 month females [14] | Mixed lymphocyte reaction assays | Source of allogeneic splenocytes for functional T cell stimulation assays |
| Recombinant LPS | Lipopolysaccharide for DC maturation [14] | DC maturation studies | Induces maturation and activation of dendritic cells |
| Antibody Panels | Anti-MHC II, CD86, CD11c, etc. [14] [116] | Flow cytometric analysis | Detection of DC maturation markers and functional states |
| CITE-seq Reagents | 127+ surface protein antibodies [116] | Multimodal single-cell profiling | Simultaneous analysis of transcriptome and surface proteome |
Broader Implications for Immune System Function
The impact of aged HSC niches extends beyond dendritic cells to affect multiple immune cell lineages and overall immune competence:
T Cell Compartment: Aging leads to decreased naïve T cell production and increased differentiation of cytotoxic T lymphocytes (CTLs) and exhausted T (Tex) cells [117]. The CD4+:CD8+ ratio typically decreases with age, and T cell receptors show reduced diversity, impairing responses to novel antigens [117] [115].
B Cell Compartment: B cell development is particularly susceptible to aging influences, with reductions in B cell precursors and alterations in B cell repertoire diversity [20] [115]. Aged B cells show decreased production of high-affinity antibodies and impaired formation of long-lived plasma cells, contributing to reduced vaccine efficacy [115].
Innate Immune Cells: Myeloid bias in aged HSCs results in expanded populations of monocytes and neutrophils, but these cells often show functional impairments in phagocytosis, efferocytosis, and pathogen clearance [20].
Tissue-Specific Effects: Recent multimodal profiling reveals that age-associated immune changes are manifested in a tissue-specific manner, with significant alterations in macrophages in mucosal sites, B cells in lymphoid organs, and circulating T cells and natural killer cells across blood and tissues [116].
The comparative analysis of young versus aged HSC niches reveals a complex landscape of microenvironmental changes that collectively drive immunosenescence. The aged niche promotes pro-inflammatory signaling, skewed differentiation toward myeloid lineages, and functional impairment of key immune cells like dendritic cells. These changes contribute to the hallmark immune deficits observed in aging: reduced response to novel pathogens, decreased vaccine efficacy, increased incidence of autoimmunity, and elevated cancer risk.
Future research directions should focus on:
- Developing more sophisticated in vitro models that better recapitulate the complexity of human bone marrow niches
- Elucidating the specific molecular mechanisms that drive niche aging
- Investigating therapeutic strategies to rejuvenate aged niches or mitigate their negative impacts on immune function
Understanding how aged HSC niches dysregulate immune cell output provides critical insights for developing interventions to restore immune competence in aging populations, potentially extending healthspan and reducing the burden of age-related diseases.
Figure 2: Experimental Workflow for Assessing Niche Effects on DCs. This diagram outlines key methodological steps for comparing young versus aged niche impacts on dendritic cell development and function [14].
Correlating Niche Alterations with Clinical Outcomes in Pre-Leukemic and Leukemic States
The bone marrow (BM) microenvironment, or niche, is a dynamic ecosystem essential for maintaining hematopoietic stem cell (HSC) homeostasis. Growing evidence indicates that alterations within this niche are not merely bystander effects but active contributors to the initiation and progression of hematological malignancies. This whitepaper synthesizes current research on how specific perturbations in the BM niche correlate with clinical outcomes in pre-leukemic conditions and overt leukemia. We detail the cellular and molecular mechanisms involved, provide quantitative data on niche-mediated prognostic factors, and outline standardized experimental protocols for investigating niche-leukemia interactions. Understanding these correlations is paramount for developing novel microenvironment-targeted therapies that could improve patient prognosis by preventing leukemic transformation and overcoming treatment resistance.
The hematopoietic stem cell (HSC) niche is a highly specialized and physiologically regulated bone marrow microenvironment that provides critical signals for the maintenance, self-renewal, and differentiation of HSCs [7] [8]. It constitutes a complex, multicellular signaling network that includes both non-hematopoietic cells (e.g., mesenchymal stem cells, osteolineage cells, endothelial cells) and hematopoietic cells (e.g., megakaryocytes, macrophages, T cells) [8] [118]. These components interact with HSCs through direct cell-to-cell contact and the secretion of cytokines, growth factors, and extracellular matrix (ECM) proteins, creating a precise balance between HSC quiescence, proliferation, and lineage commitment [8].
In the context of malignancy, this carefully balanced microenvironment can be co-opted and remodeled. Leukemic cells, particularly leukemic stem cells (LSCs), can actively transform the normal hematopoietic niche into a "leukemic niche" that supports their survival and proliferation while suppressing normal hematopoiesis [119] [118]. This remodeling occurs through bidirectional interactions where LSCs manipulate niche cells to produce factors favoring their expansion and where the altered niche, in turn, provides a sanctuary for LSCs, protecting them from chemotherapeutic insults and contributing to relapse [119] [120]. The pre-leukemic phase, characterized by conditions such as clonal hematopoiesis of indeterminate potential (CHIP), is increasingly recognized as a critical window where niche alterations may influence the probability of malignant transformation [121]. This whitepaper examines the quantitative and qualitative changes in the BM niche across the disease continuum and correlates these alterations with clinically relevant outcomes.
Quantitative Alterations in the Niche and Clinical Correlations
Specific, measurable changes in the bone marrow niche have been consistently correlated with clinical outcomes in pre-leukemic and leukemic states. The tables below summarize key quantitative alterations and their prognostic significance.
Table 1: Cellular and Molecular Alterations in the Pre-Leukemic Niche and Clinical Impact
| Alteration Type | Specific Change | Clinical Correlation/Impact | Supporting Evidence |
|---|---|---|---|
| Clonal Hematopoiesis | VAF of driver mutations >10% [121] | 39% of pre-AML individuals have clones of this size vs 4% of controls [121] | Increased risk of progression to AML [121] |
| Accumulation of ≥2 high-risk mutations (e.g., SRSF2, U2AF1, TP53, IDH1/2, RUNX1) [121] | Significantly discriminates pre-AML from benign ARCH, especially in >60-65 age group [121] | High risk for subsequent AML development [121] | |
| Inflammatory Milieu | Increased IL-1β production by myeloid cells [20] | Creates a vicious cycle of Tet2+/− clonal expansion [20] | Contributes to CHIP and increased HSPC proliferation [20] |
| Enrichment of CCL5 (RANTES) in aged microenvironment [20] | Exposure of young HSCs to Ccl5 induces myeloid bias; activates mTOR pathway [20] | Lineage skewing (myeloid bias) associated with aging and pre-malignancy [20] | |
| Niche Remodeling | Expansion of megakaryocytes and progenitors [20] | Altered spatial relationship with HSCs; potential loss of HSC quiescence [20] | Contributes to aged HSC phenotypes and inflammaging [20] |
Table 2: Niche-Mediated Mechanisms in Active Leukemia and Clinical Outcomes
| Niche Component | Leukemia-Induced Alteration | Functional Consequence | Impact on Clinical Outcome |
|---|---|---|---|
| Osteolineage Cells | Activating β-catenin mutations stimulating Jagged-1 [118] | Activation of Notch signaling in HSPCs [118] | Induction and progression of AML [118] |
| Activating Ptpn11 mutation in osteogenic progenitors [118] | Overproduction of CCL3, recruitment of monocytes [118] | Stimulation of HSC differentiation/proliferation, causing JMML [118] | |
| Vascular Niche | Aberrant activation of VEGF and IL-5 signaling in LSCs (t(8;21) AML) [122] | LSC re-entry into cell cycle via AP-1/GATA2 axis [122] | Promotion of LSC self-renewal and growth; potential driver of relapse [122] |
| Metabolic/Inflammatory | "Inflammaging" - unresolved inflammation with aging [20] | Secretion of inflammatory cytokines (e.g., IL-1, CCL5) by aged immune cells [20] | Fuels clonal expansion and selection; correlates with poor survival in older patients [20] |
| Adhesion/Protection | Niche-mediated physical protection of LSCs [119] [120] | Creation of a sanctuary from genotoxic insults like chemotherapy [119] [120] | Therapy resistance and relapse [119] [120] |
Experimental Protocols for Investigating the Niche
A multifaceted approach is required to dissect the complex interactions between leukemic cells and the bone marrow microenvironment. The following section outlines key methodologies.
Single-Cell RNA Sequencing (scRNA-Seq) of Niche Populations
Purpose: To deconvolute the cellular heterogeneity of the healthy, pre-leukemic, and leukemic bone marrow niche and identify distinct cellular states and altered transcriptional programs [20] [122].
Detailed Protocol:
- Sample Preparation: Obtain mononuclear cells from fresh human or murine bone marrow aspirates or tissue by density gradient centrifugation (e.g., using Ficoll). For leukemic samples, sort specific populations (e.g., CD34+/CD38− LSCs, CD34+/CD38+ blasts) using fluorescence-activated cell sorting (FACS) to enrich for rare subsets [122].
- Library Preparation: Use a platform such as the 10x Genomics Chromium Controller to capture single cells and barcode mRNA. Generate sequencing libraries according to the manufacturer's protocol.
- Sequencing: Perform high-depth sequencing on an Illumina platform (e.g., NovaSeq) to a recommended depth of >50,000 reads per cell.
- Bioinformatic Analysis:
- Quality Control: Filter cells based on unique molecular identifier (UMI) counts, number of genes detected, and mitochondrial gene percentage.
- Normalization and Integration: Normalize data (e.g., using SCTransform) and integrate multiple samples using tools like Harmony to remove batch effects [122].
- Clustering and Annotation: Perform principal component analysis (PCA) and graph-based clustering (e.g., Seurat, Scanpy). Annotate cell clusters using known marker genes (e.g., PECAM1 for endothelial cells, ACTA2 for mesenchymal stromal cells, MPO for myeloid blasts) [122].
- Trajectory Inference: Utilize algorithms like Monocle or PAGA to order cells along a pseudotemporal trajectory to infer differentiation paths or state transitions, such as from LSC to blast [122].
- Differential Expression: Identify marker genes for specific conditions (e.g., healthy vs. diseased HSCs, LSCs vs. blasts) to define niche-specific signatures [122].
Functional Assessment of LSC-Niche Interactions Using Patient-Derived Xenografts (PDX)
Purpose: To model human leukemia in vivo and test the functional role of specific signaling pathways in LSC maintenance and drug resistance within a physiologic microenvironment [122].
Detailed Protocol:
- Model Generation: Transplant primary human AML cells (e.g., from patient bone marrow) into immunodeficient recipient mice (e.g., NSG or NRG strains). This can be done via intravenous injection or intrafemoral injection to directly seed the BM niche.
- Perturbation Studies: Once leukemia is established (confirmed by human cell chimerism in peripheral blood or bone marrow), treat the PDX mice with:
- Small Molecule Inhibitors: For example, inhibitors targeting VEGF or IL-5 signaling to assess their effect on LSC burden [122].
- Control Vehicles: To monitor disease progression without intervention.
- Endpoint Analysis: After a defined treatment period, sacrifice the mice and analyze the bone marrow.
- Flow Cytometry: Quantify the frequency of human LSCs (e.g., CD34+/CD38−) and blasts.
- Colony-Forming Unit (CFU) Assays: Plate sorted LSCs in methylcellulose to assess their self-renewal and proliferative capacity ex vivo [122].
- Secondary Transplantation: Inject BM cells from primary PDX mice into secondary recipient mice to rigorously assess the long-term repopulating potential and self-renewal of LSCs after in vivo perturbation [120].
3DIn VitroNiches for Drug Screening
Purpose: To create a biomimetic human bone marrow microenvironment for high-throughput drug testing and mechanistic studies, reducing reliance on animal models [7].
Detailed Protocol:
- Scaffold Selection: Choose a suitable 3D scaffold, such as:
- Biomimetic Hydrogel: A matrix composed of collagen, hyaluronic acid, or other ECM components to mimic the physical and biochemical properties of the BM [7].
- Bone Marrow-on-a-Chip: A microfluidic device lined with BM endothelial and stromal cells to recreate the vascular niche and dynamic fluid flow [7].
- Model Population: Seed the 3D scaffold with:
- Stromal Components: Human mesenchymal stem cells (MSCs), osteoprecursors, and endothelial cells.
- Target Cells: Healthy HSPCs, pre-leukemic cells, or primary LSCs.
- Intervention and Readout:
- Drug Exposure: Treat the 3D culture with chemotherapeutic agents or targeted drugs.
- Functional Assays: Measure outcomes such as LSC viability (via ATP-based assays), apoptosis (by caspase activation), proliferation (by Ki67 staining or EdU incorporation), and differentiation (by flow cytometry for lineage markers) [7].
Visualization of Key Signaling Pathways
The following diagrams, generated using Graphviz DOT language, illustrate critical signaling pathways involved in niche-mediated support of leukemic stem cells.
Diagram Title: VEGF/IL-5 Signaling Circuit in t(8;21) LSCs
This diagram illustrates the aberrant activation of VEGF and IL-5 signaling pathways in t(8;21) Acute Myeloid Leukemia (AML) LSCs, as identified in recent studies [122]. The pathways form a regulatory circuit with the driver oncoprotein RUNX1::ETO and an AP-1/GATA2 axis, enabling LSCs to re-enter the cell cycle while preserving self-renewal capacity, a key mechanism for relapse.
The Scientist's Toolkit: Essential Research Reagents and Models
Advancing research in the leukemic niche requires a specific toolkit of reagents, models, and technologies. The following table details key solutions for investigating niche alterations.
Table 3: Essential Research Reagents and Models for Leukemic Niche Studies
| Tool Category | Specific Item/Model | Key Function/Application | Reference |
|---|---|---|---|
| In Vivo Models | Patient-Derived Xenograft (PDX) Mice (e.g., NSG, NRG) | Models human leukemia in a physiologic microenvironment; essential for studying LSC biology and therapy resistance. | [122] |
| Heterotopic Ossicle Model | Recapitulates humanized bone marrow microenvironment with human cells and ECM components for niche studies. | [7] | |
| In Vitro Models | 3D Biomimetic Hydrogel Scaffolds | Provides 3D architecture for long-term HSC/LSC expansion and differentiation in vitro. | [7] |
| Bone Marrow-on-a-Chip (Microfluidic Device) | Recreates dynamic vascular niche for disease modeling and high-throughput drug screening. | [7] | |
| Cell Isolation Tools | Fluorescence-Activated Cell Sorting (FACS) Panels (e.g., CD34, CD38, CD123, CD96) | Isolation of highly purified populations of HSCs, LSCs, and niche cells for functional analysis. | [119] [122] |
| Targeting Agents | Small Molecule Inhibitors (e.g., targeting VEGF, IL-5, JAK/STAT pathways) | Functional perturbation of specific signaling pathways identified as critical for LSC maintenance. | [122] |
| CXCR4 Antagonists (e.g., AMD3100/Plerixafor) | Blocks CXCR4/SDF-1 interaction; used to mobilize HSCs and potentially disrupt LSC niche protection. | [118] | |
| Analytical Technologies | Single-Cell RNA Sequencing (scRNA-Seq) | Unravels cellular heterogeneity and identifies novel cellular states in the niche and leukemic populations. | [20] [122] |
| ATAC-Seq / DNaseI-Seq | Maps genome-wide chromatin accessibility to define regulatory landscapes in LSCs vs. blasts. | [122] | |
| Advanced Microscopy (e.g., Multiphoton, Confocal) | Enables spatial analysis of cell localization and interactions within the native bone marrow niche. | [7] [20] |
The correlation between bone marrow niche alterations and clinical outcomes in pre-leukemic and leukemic states is unequivocal. The transition from a health-maintaining to a malignancy-supporting microenvironment involves quantifiable changes in cellular composition, signaling pathways, and physical properties. Key alterations, such as the expansion of inflammatory immune cells, the aberrant activation of developmental signaling pathways like VEGF and IL-5 in LSCs, and the physical protection of LSCs within the niche, are strongly linked to disease initiation, therapy resistance, and relapse.
Future research must focus on translating this mechanistic understanding into clinical applications. This includes:
- Biomarker Development: Validating niche-derived factors or cellular alterations as prognostic biomarkers to identify high-risk CHIP or early leukemia.
- Therapeutic Targeting: Developing and clinically testing niche-targeting agents, used in combination with conventional therapies, to disrupt the protective sanctuary and eradicate LSCs.
- Niche Repair Strategies: Exploring approaches to restore normal niche function after chemotherapy or transplantation to improve hematopoietic reconstitution and reduce relapse.
The ongoing development of sophisticated in vitro and in vivo models, coupled with high-resolution omics technologies, will continue to refine our understanding of these dynamic interactions, ultimately paving the way for more effective and curative strategies for hematological malignancies.
Conclusion
The hematopoietic stem cell niche is far from a passive scaffold; it is a dynamic, instructible entity that is fundamental to health, aging, and disease. Synthesizing key insights reveals that age-related and inflammatory remodeling of the niche actively contributes to hematopoietic decline and the initiation of blood cancers. The development of sophisticated in vitro models now provides unprecedented opportunities to dissect these mechanisms and perform high-throughput therapeutic discovery. Crucially, the niche itself presents a compelling and novel therapeutic target. Future research must focus on longitudinal studies to understand the 'memory' of a diseased niche, the development of combinatorial therapies that target both malignant cells and their supportive microenvironment, and the translation of niche-modulating strategies into clinical practice to prevent disease progression and improve regenerative medicine outcomes.
References
- 1. Stem Cell Niche Concept: Search for Current Expert ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12429054/]
- 2. Making sense of hematopoietic stem cell niches - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4408288/]
- 3. Hematopoietic Stem Cells and Their Niche in Bone Marrow [https://www.mdpi.com/1422-0067/25/13/6837]
- 4. Single cell analysis of the localization of the hematopoietic ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1166758/full]
- 5. Haematopoietic stem cell number is not solely defined by ... [https://www.nature.com/articles/s41586-025-09462-5]
- 6. Hematopoietic Stem Cell Niches and Signals Controlling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7726109/]
- 7. Reconstruction of hematopoietic stem cell niches in vitro [https://pmc.ncbi.nlm.nih.gov/articles/PMC12648611/]
- 8. Understanding the Bone Marrow Niche: A Key to ... [https://blog.crownbio.com/understanding-the-bone-marrow-niche]
- 9. Bone marrow niches for hematopoietic stem cells in ... [https://www.sciencedirect.com/science/article/pii/S0301472X25000402]
- 10. Exploring MSC and HSPC interactions: new frontiers in ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04753-0]
- 11. Hematopoietic stem cell size heterogeneity is not linked to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12129979/]
- 12. Interactions of Hematopoietic and Associated Mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12469976/]
- 13. Unraveling hematopoietic stem cell ontogeny through ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1704887/full]
- 14. Differential effects of young and old hematopoietic stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224859/]
- 15. Hematopoietic stem cell metabolism within the bone marrow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11755706/]
- 16. Metabolic Plasticity and Hematopoietic Stem Cell Biology [https://pmc.ncbi.nlm.nih.gov/articles/PMC3736335/]
- 17. Hypoxia inducible factor 1α-driven steroidogenesis impacts ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00777-9]
- 18. Interactions of Hematopoietic and Associated ... [https://pubmed.ncbi.nlm.nih.gov/41009600/]
- 19. Inflammation rewires bone marrow microenvironment long ... [https://www.embl.org/news/science-technology/inflammation-rewires-bone-marrow-microenvironment-long-before-leukaemias-develop/]
- 20. The aging hematopoietic stem cell niche: a mini review [https://www.frontiersin.org/journals/hematology/articles/10.3389/frhem.2025.1525132/full]
- 21. Is the bone marrow microenvironment the hidden catalyst ... [https://www.nature.com/articles/s41375-025-02630-6]
- 22. Role of Myeloid Cells in Age-Related Diseases - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC8760106/]
- 23. Inflammation and aging: signaling pathways and ... [https://www.nature.com/articles/s41392-023-01502-8]
- 24. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5107568/]
- 25. Hematopoietic Stem Cell Aging: Mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12649273/]
- 26. Mechanisms and rejuvenation strategies for aged ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-020-00864-8]
- 27. Aging is associated with functional and molecular changes ... [https://www.nature.com/articles/s41467-024-52318-1]
- 28. Reconstruction of Hematopoietic Stem Cell Niches In Vitro [https://www.sciencedirect.com/science/article/pii/S259000642501107X]
- 29. Clonal hematopoiesis: from mechanisms to clinical intervention [https://pmc.ncbi.nlm.nih.gov/articles/PMC8854454/]
- 30. Clonal hematopoiesis [https://en.wikipedia.org/wiki/Clonal_hematopoiesis]
- 31. Clonal hematopoiesis and inflammation: A review of ... [https://www.sciencedirect.com/science/article/abs/pii/S1040842823002755]
- 32. Clonal hematopoiesis as a model for premalignant ... [https://www.sciencedirect.com/science/article/pii/S0301472X19311336]
- 33. The aging hematopoietic stem cell niche: Phenotypic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5244432/]
- 34. Restoring bone marrow niche function rejuvenates aged ... [https://www.nature.com/articles/s41467-023-37783-4]
- 35. Differential effects of young and old hematopoietic stem cell ... [https://immunityageing.biomedcentral.com/articles/10.1186/s12979-025-00517-9]
- 36. HSC Niche Dynamics in Regeneration, Pre-malignancy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10020982/]
- 37. Modeling the Bone Marrow Microenvironment to Better ... [https://pubmed.ncbi.nlm.nih.gov/40805267/]
- 38. Translational Advances in the 3D Modeling of Blood Cancers [https://www.mdpi.com/2673-7523/5/4/51]
- 39. Bioengineered niches that recreate physiological ... [https://www.nature.com/articles/s41467-024-50054-0]
- 40. Bone marrow organoid-based platforms overcoming barriers ... [https://j-organoid.org/m/journal/view.php?number=78]
- 41. Functionalized 3D scaffolds for engineering the ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.968086/full]
- 42. Biomimetic artificial bone marrow niches for the scale up of ... [https://www.sciencedirect.com/science/article/pii/S0142961225005319]
- 43. AACR 2025: Crown Bioscience Unveils 3D Bone Marrow ... [https://www.crownbio.com/about-us/news-and-events/aacr-2025-crown-bioscience-unveils-3d-bone-marrow-niche-in-vitro-models-for-oncology-preclinical-screening-in-hematological-malignancies]
- 44. First-of-its-kind 3D bone marrow model [https://www.selectscience.net/article/first-of-its-kind-3d-bone-marrow-model-advances-blood-cancer-research]
- 45. 3D Bone Marrow Models: Transforming Drug Discovery for ... [https://www.crownbio.com/3d-bone-marrow-models]
- 46. Interactions of Hematopoietic Stem Cells with Bone Marrow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9423787/]
- 47. Complexities of modeling the bone marrow ... [https://www.sciencedirect.com/science/article/pii/S0301472X24000924]
- 48. Using Organ-on-a-Chip Technology to Unlock Patient ... [https://emulatebio.com/using-organ-on-a-chip-technology-to-unlock-patient-derived-precision-medicine/]
- 49. Generation of complex bone marrow organoids from ... [https://www.nature.com/articles/s41592-024-02172-2]
- 50. A microfluidic bone marrow chip for the safety profiling of ... [https://www.nature.com/articles/s42003-025-08137-1]
- 51. High-Throughput Microfluidic Generation of Self ... [https://www.sciencedirect.com/science/article/pii/S266683192500133X]
- 52. The haematopoietic stem cell niche at a glance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3215569/]
- 53. Architectural Design of Biomaterial Scaffolds for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599877/]
- 54. The physical microenvironment of hematopoietic stem cells ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-019-1422-7]
- 55. Current insights into the bone marrow niche: From biology ... [https://www.sciencedirect.com/science/article/pii/S0142961222002083]
- 56. Additive-Free Hyaluronic Acid-Based Bioink for 3D ... [https://www.sciencedirect.com/science/article/pii/S259000642501083X]
- 57. Living Heart Tissue Created with 3D Printing Technology [https://hospitalnews.com/researchers-create-living-heart-tissue-with-a-3d-printer-for-testing-new-treatments/]
- 58. Bioengineered 3D in vitro Human Bone Marrow Niche [https://www.crick.ac.uk/careers-study/vacancies/2025-09-23-bonnet-lab-bioengineered-3d-in-vitro-human-bone-marrow-niche]
- 59. Reconstruction of hematopoietic stem cell niches in vitro [https://pubmed.ncbi.nlm.nih.gov/41312135/]
- 60. Long-term engrafting multilineage hematopoietic cells ... [https://www.nature.com/articles/s41587-024-02360-7]
- 61. High throughput cell-based assay of hematopoietic ... [https://pubmed.ncbi.nlm.nih.gov/11788056/]
- 62. Series GSE301224 - GEO Accession viewer - NIH [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE301224]
- 63. Rhosin shown to reverse blood stem cell ageing [https://www.drugtargetreview.com/news/190920/rhosin-shown-to-reverse-blood-stem-cell-ageing/]
- 64. Hematopoietic Stem Cells (HSCs) [https://www.thermofisher.com/br/en/home/life-science/stem-cell-research/hematopoietic-stem-cells.html]
- 65. Hematopoietic Stem Cells and Their Niche in Bone Marrow [https://pmc.ncbi.nlm.nih.gov/articles/PMC11241602/]
- 66. Artificial intelligence and systems biology analysis in stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12476622/]
- 67. Imaging methods used to study mouse and human HSC ... [https://www.sciencedirect.com/science/article/abs/pii/S8756328218301765]
- 68. Advanced Microscopy Technique Offers a New Look Inside ... [https://medicine.yale.edu/news-article/advanced-microscopy-technique-offers-a-new-look-inside-cells/]
- 69. Advanced Microscopy Techniques for Molecular Biophysics [https://pmc.ncbi.nlm.nih.gov/articles/PMC10298208/]
- 70. Machine learning–based scoring models to predict ... [https://www.sciencedirect.com/science/article/pii/S2473952921006170]
- 71. CHIP: a clonal odyssey of the bone marrow niche - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11290965/]
- 72. Inflammatory stromal and T cells mediate human bone ... [https://www.nature.com/articles/s41467-025-65803-y]
- 73. Bone marrow microenvironment in myelodysplastic neoplasms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12065391/]
- 74. Inflammation rewires the bone marrow microenvironment long ... [https://biomedizin.unibas.ch/en/communication/details/inflammation-rewires-the-bone-marrow-microenvironment-long-before-leukemias-develop-zaugg-lab/]
- 75. Inflammation turns bone marrow into a breeding ground for ... [https://www.sciencedaily.com/releases/2025/11/251118220049.htm]
- 76. Cellular crosstalk in the bone marrow niche [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05900-6]
- 77. Hematopoietic Stem Cell Niches and Signals Controlling ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.600127/full]
- 78. Comprehensive characterization of human bone marrow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12580717/]
- 79. Mesenchymal niche-specific expression of Cxcl12 controls ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6499704/]
- 80. Ebf3+ niche-derived CXCL12 is required for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10600098/]
- 81. Aging of the Hematopoietic Stem Cell Niche: New Tools to ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.738204/full]
- 82. Cancer Cell Dormancy in the Bone Microenvironment - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528268/]
- 83. Mechanisms and implications of cancer cell dormancy in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7030181/]
- 84. Cancer cell dormancy: An update to 2025 [https://bmrat.org/index.php/BMRAT/article/view/994]
- 85. Nature 揭秘肿瘤细胞“休眠”机制,有效预防转移癌 [https://news.bioon.com/article/6787913.html]
- 86. Harnessing the Stem Cell Niche in Regenerative Medicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC10816024/]
- 87. Modulating the stem cell niche for tissue regeneration - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4422171/]
- 88. Bone Marrow Niche Attenuation by Non-Steroidal Anti- ... [https://www.sciencedirect.com/science/article/pii/S0006497119586566]
- 89. Catch me if you can: how AML and its niche escape ... [https://www.nature.com/articles/s41375-021-01350-x]
- 90. Inflammation in cancer: therapeutic opportunities from new ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02243-8]
- 91. Challenges and innovations in hematopoietic stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11362137/]
- 92. The quest for the holy grail: overcoming challenges in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10345135/]
- 93. In Vitro to In Vivo Translation in Lead Optimization [https://www.sygnaturediscovery.com/news-and-events/blog/in-vitro-to-in-vivo-translation-in-lead-optimization/]
- 94. Visualizing hematopoiesis as a stochastic process - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6199667/]
- 95. Visualizing hematopoiesis as a stochastic process [https://www.sciencedirect.com/science/article/abs/pii/S2473952920308211]
- 96. Stem Cell Niche Concept: Search for Current Expert ... [https://www.mdpi.com/1422-0067/26/17/8422]
- 97. Aged hematopoietic stem cells are refractory to bloodborne ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8155813/]
- 98. Bone marrow niches for hematopoietic stem cells: life span ... [https://www.sciencedirect.com/science/article/abs/pii/S0006497124009340]
- 99. A non-genotoxic stem cell therapy boosts lymphopoiesis ... [https://www.nature.com/articles/s41467-025-60464-3]
- 100. enhancing rejuvenation and accelerating aging [https://www.jci.org/articles/view/123946]
- 101. Stem Cell Niche Concept: Search for Current Expert ... [https://pubmed.ncbi.nlm.nih.gov/40943342/?utm_source=SimplePie&utm_medium=rss&utm_campaign=pubmed-2&utm_content=1pq-4TZ0w1pIimA2HHLJZYTkSjaU335U-aRSFRxJ31Blahz9sN&fc=20220524061305&ff=20250916055026&v=2.18.0.post9+e462414]
- 102. Myocardial infarction activates CCR2+ hematopoietic stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4426344/]
- 103. dependent activation of hematopoietic stem and progenitor ... [https://link.springer.com/article/10.1007/s00395-022-00927-6]
- 104. Bone marrow niches for hematopoietic stem cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11289431/]
- 105. Next generation lineage tracing and its applications ... [https://www.nature.com/articles/s41540-025-00542-w]
- 106. Single-cell lineage tracing techniques in hematology [https://pmc.ncbi.nlm.nih.gov/articles/PMC11877926/]
- 107. Advancements in Hematopoietic Stem Cell Therapy [https://www.mdpi.com/2813-9909/2/4/18]
- 108. Single Cell Spatial Transcriptomics Reveals Immunotherapy ... [https://pubmed.ncbi.nlm.nih.gov/39975227/]
- 109. Characterization of the bone marrow architecture ... [https://www.nature.com/articles/s42003-025-08975-z]
- 110. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 111. An overview of single-cell RNA sequencing and spatial ... [https://frontlinegenomics.com/an-overview-of-single-cell-rna-sequencing-and-spatial-transcriptomics/]
- 112. Advances in spatial transcriptomics and related data analysis ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-023-04150-2]
- 113. Systematic comparison of sequencing-based spatial ... [https://www.nature.com/articles/s41592-024-02325-3]
- 114. Frontiers | Deciphering the bone ’s role in... marrow microenvironment [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1613265/full]
- 115. Mechanistic insights and biomarker discovery in immune ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1637191/full]
- 116. Multimodal profiling reveals tissue-directed signatures of ... [https://www.nature.com/articles/s41590-025-02241-4]
- 117. Comprehensive characterisation of age-related changes in ... [https://www.nature.com/articles/s41419-025-08007-y]
- 118. Hematopoietic Stem Cell Niche During Homeostasis, ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.621214/full]
- 119. Leukemic Stem Cells: From Leukemic Niche Biology to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8554324/]
- 120. The leukemic stem cell niche - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC2723012/]
- 121. Evolutionary trajectory of leukemic clones and its clinical ... [https://haematologica.org/article/view/8890]
- 122. Leukemic stem cells activate lineage inappropriate ... [https://www.nature.com/articles/s41467-024-45691-4]