Cryopreservation of MSC-Based Tissue-Engineered Structures: Protocols, Challenges, and Clinical Translation
This comprehensive review addresses the critical process of cryopreserving mesenchymal stem cell (MSC)-based tissue-engineered structures, a pivotal step for clinical translation in regenerative medicine.
Cryopreservation of MSC-Based Tissue-Engineered Structures: Protocols, Challenges, and Clinical Translation
Abstract
This comprehensive review addresses the critical process of cryopreserving mesenchymal stem cell (MSC)-based tissue-engineered structures, a pivotal step for clinical translation in regenerative medicine. Covering foundational principles to advanced applications, we explore the dual challenge of maintaining both cell viability and complex structural integrity post-thaw. The article details established slow-freezing and vitrification methodologies, analyzes their impacts on MSC functionality including immunomodulatory properties and differentiation potential, and provides troubleshooting strategies for common limitations like cryoprotectant toxicity and ice crystal formation. With a focus on validation frameworks and comparative efficacy, this resource equips researchers and drug development professionals with the knowledge to advance cryopreserved MSC products from the laboratory to clinical implementation for treating hematological diseases, orthopedic conditions, and other therapeutic applications.
MSC Biology and Cryobiology Fundamentals for Tissue Engineering
In regenerative medicine and tissue engineering, Mesenchymal Stromal Cells (MSCs) represent a cornerstone for therapeutic applications. The minimal criteria for defining MSCs were established in 2006 by the International Society for Cellular Therapy (ISCT) and consist of three fundamental pillars: (1) adherence to plastic under standard culture conditions; (2) expression of specific surface markers (CD105, CD73, CD90) and lack of expression of hematopoietic markers; and (3) capacity for trilineage differentiation into osteocytes, adipocytes, and chondrocytes in vitro [1] [2] [3]. These criteria provide the essential framework for qualifying MSC populations across different tissue sources, ensuring consistency and reliability in research and clinical applications. Within the context of cryopreservation research for tissue-engineered structures, a rigorous assessment of these defining characteristics post-thaw is paramount. The process of freezing and thawing can significantly impact MSC viability, functionality, and differentiation potential, making pre- and post-cryopreservation characterization a critical step in developing off-the-shelf cellular therapeutics for bone, cartilage, and adipose tissue regeneration [4].
Core Defining Criteria: Experimental Assessment and Protocols
Plastic Adherence
Principle: The plastic-adherent property is a functional characteristic that serves as the primary, straightforward method for isolating MSCs from heterogeneous tissue digests. This adherence under standard culture conditions forms the basis for their expansion in vitro [1] [5].
Experimental Protocol: Isolation and Expansion of Plastic-Adherent MSCs
- Sample Preparation: Isolate mononuclear cells from the tissue of interest (e.g., bone marrow, adipose tissue) using enzymatic digestion (e.g., Collagenase P) and/or density gradient centrifugation (e.g., Ficoll-Paque) [3].
- Seeding: Plate the obtained cell suspension in standard tissue culture flasks using a complete medium, typically consisting of αMEM or DMEM, supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin [6] [3].
- Incubation and Adherence: Culture the cells in a humidified incubator at 37°C with 5% CO₂.
- Removal of Non-Adherent Cells: After 48-72 hours, carefully remove the culture medium, which contains non-adherent cells (primarily hematopoietic lineages). Wash the adherent layer gently with phosphate-buffered saline (PBS) to remove any residual non-adherent cells.
- Expansion: Continue to culture the adherent, fibroblast-like cells, refreshing the medium every 2-3 days until the cells reach 70-80% confluence. These can then be passaged using a cell dissociation reagent like Accutase or Trypsin-EDTA [6] [3].
Surface Marker Expression
Principle: The ISCT defines a specific immunophenotype for MSCs. A population must show ≥95% expression of CD105, CD73, and CD90, and ≤2% expression of hematopoietic markers (CD45, CD34, CD14 or CD11b, CD79α or CD19, and HLA-DR) [1] [5]. It is critical to note that marker expression can be altered by in vitro culture conditions and may not always reflect the in vivo phenotype [6].
Experimental Protocol: Immunophenotyping by Flow Cytometry
- Cell Preparation: Harvest MSCs (preferably at early passages, e.g., P3-P5) and create a single-cell suspension. Wash the cells with a flow cytometry staining buffer (PBS with 2% FBS).
- Antibody Staining: Aliquot cells into tubes and incubate with fluorochrome-conjugated antibodies against the target proteins (CD105, CD73, CD90) and the negative markers (CD45, CD34, etc.), along with appropriate isotype controls, for 30 minutes at 4°C in the dark.
- Washing and Fixation: Wash the cells twice with staining buffer to remove unbound antibody. The cells can be fixed with a 1-4% paraformaldehyde solution if not analyzed immediately.
- Data Acquisition and Analysis: Acquire the stained cells on a flow cytometer. Analyze the data to determine the percentage of positively stained cells in the population, gating on the viable cell population based on forward/side scatter or a viability dye [6] [2].
Table 1: Key Surface Markers for MSC Definition
| Marker | Expression | Function | ISCT Requirement |
|---|---|---|---|
| CD105 (Endoglin) | Positive | Receptor for TGF-β; essential for angiogenesis [1]. | ≥95% |
| CD73 | Positive | Ecto-5'-nucleotidase; catalyzes AMP to adenosine [1]. | ≥95% |
| CD90 (Thy-1) | Positive | GPI-anchored protein; mediates cell-cell and cell-ECM interactions [1]. | ≥95% |
| CD45 | Negative | Protein tyrosine phosphatase; marker for all leukocytes [1]. | ≤2% |
| CD34 | Negative | Cell adhesion factor; marker for hematopoietic stem cells [1]. | ≤2% |
| HLA-DR | Negative | MHC Class II molecule; indicates an activated, immunogenic state [1]. | ≤2% |
Trilineage Differentiation Potential
Principle: The multipotency of MSCs is functionally validated by their ability to differentiate into osteoblasts, adipocytes, and chondrocytes under specific in vitro inductive conditions. This confirms their "stem" character and utility in tissue engineering.
Experimental Protocol: Trilineage Differentiation and Analysis
1. Osteogenic Differentiation
- Induction: Culture MSCs at confluence in osteogenic medium (e.g., basal medium supplemented with 50 µg/mL ascorbate-2-phosphate, 10 mM β-glycerophosphate, and 10⁻⁸ M dexamethasone) for 21-28 days, with medium changes every 3-4 days [6] [7].
- Analysis:
- Staining: Fix cells and perform Von Kossa staining to detect mineralized calcium phosphate deposits (stains black) [6].
- Quantitative Assay: Use Alizarin Red S staining to quantify calcium deposition. The bound dye can be eluted and measured spectrophotometrically.
2. Adipogenic Differentiation
- Induction: Culture confluent MSCs in adipogenic induction medium (e.g., containing 1 µM dexamethasone, 0.5 mM isobutylmethylxanthine, 10 µg/mL insulin, and 200 µM indomethacin) for 14-21 days, with regular medium changes [7].
- Analysis:
- Staining: Fix cells and stain with Oil Red O to visualize intracellular lipid droplets (stains red) [7].
- Quantitative Assay: Elute the bound Oil Red O dye with isopropanol and measure the absorbance.
3. Chondrogenic Differentiation
- Induction: Pellet 2.5 x 10⁵ MSCs in a conical tube and culture in chondrogenic medium (e.g., high-glucose DMEM with 1% ITS+1, 50 µg/mL ascorbate-2-phosphate, 100 nM dexamethasone, and 10 ng/mL TGF-β3) for 21-28 days [6].
- Analysis:
- Staining: Embed the pellet in paraffin, section it, and perform Alcian Blue staining to detect sulfated proteoglycans in the extracellular matrix (stains blue) [6].
- Histology: Safranin-O staining can also be used to assess cartilage matrix production.
Table 2: Standardized Protocols for Trilineage Differentiation
| Lineage | Induction Media Key Components | Culture Duration | Key Staining Markers | Critical Factors |
|---|---|---|---|---|
| Osteogenic | Ascorbate-2-phosphate, β-glycerophosphate, Dexamethasone [6] | 21-28 days | Von Kossa, Alizarin Red S | High cell density at induction is crucial. |
| Adipogenic | Dexamethasone, IBMX, Insulin, Indomethacin [7] | 14-21 days | Oil Red O | Cyclic induction/maintenance protocols are often used. |
| Chondrogenic | TGF-β3, Dexamethasone, Ascorbate-2-phosphate, ITS+1 [6] | 21-28 days | Alcian Blue, Safranin-O | 3D pellet or micromass culture is required. |
The following workflow summarizes the key experimental steps for defining MSCs according to the ISCT criteria, from isolation to final characterization.
Molecular Regulation of MSC Stemness and Differentiation
The core properties of MSCs are governed by a complex network of intrinsic genetic and epigenetic regulators. Understanding this molecular basis is essential for controlling their stemness and differentiation potential, particularly after the stress of cryopreservation.
Key transcriptional factors include the Twist family (Twist1/Twist2), which promote proliferation and inhibit osteogenesis to maintain stemness, partly by repressing senescence genes like p16 [7]. SOX2 is another crucial factor whose reduced expression is linked to MSC senescence during in vitro expansion [7]. The OCT4 transcription factor, particularly the OCT4A isoform, enhances proliferation, colony formation, and chondrogenesis, and its knockdown suppresses adipogenesis [7]. Furthermore, the HOX family of genes provides a stable "HOX code" that reflects the tissue origin of MSCs and regulates their specific differentiation capacities; for example, HOXA5 promotes osteogenic differentiation [7].
The differentiation process is also heavily influenced by external mechanical cues, a field known as mechanobiology. MSCs sense the rigidity of their substrate via focal adhesions and force generation by the actin cytoskeleton [8]. Stiffer matrices promote osteogenesis through increased ROCK, FAK, and ERK signaling, while softer matrices favor adipogenesis and chondrogenesis [8]. The following diagram illustrates the key molecular pathways that regulate MSC stemness and lineage commitment.
The Scientist's Toolkit: Essential Research Reagent Solutions
A standardized set of reagents and materials is fundamental for the reproducible isolation, expansion, and characterization of MSCs.
Table 3: Key Research Reagents for MSC Work
| Category / Reagent | Specific Example | Function in MSC Research |
|---|---|---|
| Digestive Enzymes | Collagenase P [6] | Digests extracellular matrix in tissues (e.g., periosteum, cartilage) to isolate individual cells. |
| Culture Media | αMEM / DMEM + 10% FBS [6] [3] | Standard basal medium for MSC expansion and maintenance. |
| Cell Dissociation Reagents | Accutase, Trypsin-EDTA [6] [3] | Detaches adherent MSCs from culture plastic for passaging or analysis. |
| Flow Cytometry Antibodies | Anti-CD105, -CD73, -CD90, -CD45, -CD34 [6] [2] | Critical for immunophenotyping and confirming MSC identity per ISCT criteria. |
| Osteogenic Inducers | Ascorbate-2-phosphate, β-glycerophosphate, Dexamethasone [6] | Key components in osteogenic differentiation media to induce bone formation. |
| Adipogenic Inducers | Insulin, IBMX, Indomethacin, Dexamethasone [7] | Key components in adipogenic differentiation media to induce fat formation. |
| Chondrogenic Inducers | TGF-β3, ITS+1 Supplement, Ascorbate-2-phosphate [6] | Key components in chondrogenic differentiation media to induce cartilage formation. |
| Histological Stains | Alizarin Red S, Oil Red O, Alcian Blue [6] [7] | Used to visually confirm successful trilineage differentiation. |
| Cryoprotective Agents (CPA) | Dimethyl Sulfoxide (DMSO) [4] | Permeable CPA used in slow-freezing protocols to protect cells from ice crystal damage. |
Critical Considerations in the Context of Cryopreservation
The process of cryopreserving and thawing MSCs presents unique challenges that can impact the very criteria used to define them. Researchers must be aware of these factors to ensure the quality of their cell products.
- Impact on Viability and Recovery: The standard slow-freezing method, while simple and widely used, typically results in ~70-80% post-thaw cell survival [4]. The choice of cryoprotective agent (CPA) is critical; DMSO is effective but can be cytotoxic and must be thoroughly washed from the cells post-thaw to avoid adverse effects in clinical applications [4].
- Alteration of Surface Markers: Cryopreservation can potentially affect the expression of key surface markers. While markers like CD73 and CD90 are generally retained, studies show that differentiation can induce changes, such as the loss of CD106 and CD146 during osteogenesis [6]. Therefore, immunophenotyping should be performed on recovered cells post-thaw to confirm identity.
- Functional Competence Post-Thaw: The most critical test for cryopreserved MSCs intended for tissue engineering is the retention of their trilineage differentiation potential. The stress of freezing and thawing can impair this fundamental capacity. It is therefore a mandatory quality control check to perform differentiation assays on cells that have been cryopreserved and recovered, rather than assuming functionality based on pre-freeze data [4].
- Cryopreservation Methods: The two main methods are slow freezing (controlled-rate cooling to -80°C followed by liquid nitrogen storage) and vitrification (ultra-rapid cooling using high CPA concentrations). While slow freezing is the current standard for MSCs due to its practicality, vitrification is an area of active development to potentially improve outcomes [4].
The rigorous application of the ISCT's defining criteria—plastic adherence, specific surface marker expression, and trilineage differentiation—forms the bedrock of credible MSC research. For the field of cryopreservation in tissue engineering, these criteria are not merely initial characterization steps but are essential quality control metrics that must be validated post-thaw. A deep understanding of the molecular regulators of MSC stemness and differentiation, combined with a standardized toolkit of reagents and protocols, enables researchers to reliably isolate, characterize, and preserve functional MSCs. This ensures that these versatile cells retain their therapeutic potential, paving the way for the development of effective, off-the-shelf, MSC-based tissue-engineered structures.
Mesenchymal stem cells (MSCs) have emerged as a highly promising tool in regenerative medicine due to their self-renewal capacity, multi-lineage differentiation potential, and immunomodulatory properties [1]. These cells can be isolated from a wide variety of tissues, but selecting the optimal source is critical for specific tissue engineering applications. This Application Note provides a systematic comparison of MSCs derived from four key sources—bone marrow, adipose tissue, umbilical cord, and amnion—with a specific focus on their application in developing cryopreserved tissue-engineered structures. The standardization of MSC sources is fundamental for creating off-the-shelf products that maintain consistent viability, functionality, and therapeutic potential post-thaw, thereby advancing their clinical translation [9].
The biological characteristics of MSCs vary significantly depending on their tissue of origin. These differences influence their proliferation capacity, differentiation potential, senescence, and paracrine activity, which collectively determine their suitability for specific tissue engineering applications.
Growth Characteristics and Senescence
Table 1: Growth Characteristics and Senescence Markers of Different MSC Sources
| MSC Source | Proliferation Capacity | Population Doubling Time | Cumulative Population Doublings | Senescence Markers (p53, p21, p16) | Clonality (CFU-F Assay) |
|---|---|---|---|---|---|
| Bone Marrow | Moderate [10] | ~30-40 hours [10] | Moderate [10] | High expression with culture expansion [10] | 16.5 ± 4.4 [10] |
| Adipose Tissue | Lower than BM and UCB [10] | Longer than UCB-MSCs [10] | Significantly less than BM and UCB [10] | High expression with culture expansion [10] | 6.4 ± 1.6 [10] |
| Umbilical Cord | Highest among sources [11] [10] | Shortest [10] | Highest [10] | Significantly lower expression [11] [10] | 23.7 ± 5.8 [10] |
| Amnion | Higher than UC-MSCs in some studies [12] | Information not specified in search results | Information not specified in search results | Information not specified in search results | Information not specified in search results |
Differentiation Potential and Immunophenotype
Table 2: Differentiation Potential and Molecular Profiles of MSC Sources
| MSC Source | Osteogenic Potential | Chondrogenic Potential | Adipogenic Potential | Immunomodulatory Capacity | Key Molecular Features |
|---|---|---|---|---|---|
| Bone Marrow | High [13] | High [13] | High [13] | Significantly inhibits T-cell proliferation; high IL10 and TGFB1 [13] | DLX5 expression associated with osteogenic potential [13] |
| Adipose Tissue | High [13] [14] | High [13] [14] | High [13] [14] | Similar immunophenotype to other sources [14] | Shares gene expression profile with BM-MSCs [13] |
| Umbilical Cord | Variable, lower than BM [11] | Enhanced vs. BM [11] | Lower than BM [11] | Similar immunophenotype to other sources [14] | Higher expression of tenogenic genes (MMP3, SCX, DCN, TNC) [11] |
| Amnion | Higher efficiency in serum-free conditions [12] | Similar to UC and CP [12] | Lower efficiency [12] | Information not specified in search results | Unique gene expression profile under serum-free conditions [12] |
All MSC sources express standard positive markers (CD105, CD73, CD90) and lack hematopoietic markers (CD45, CD34, CD14/CD11b, CD79α/CD19, HLA-DR) according to International Society for Cellular Therapy (ISCT) criteria [5] [4] [1].
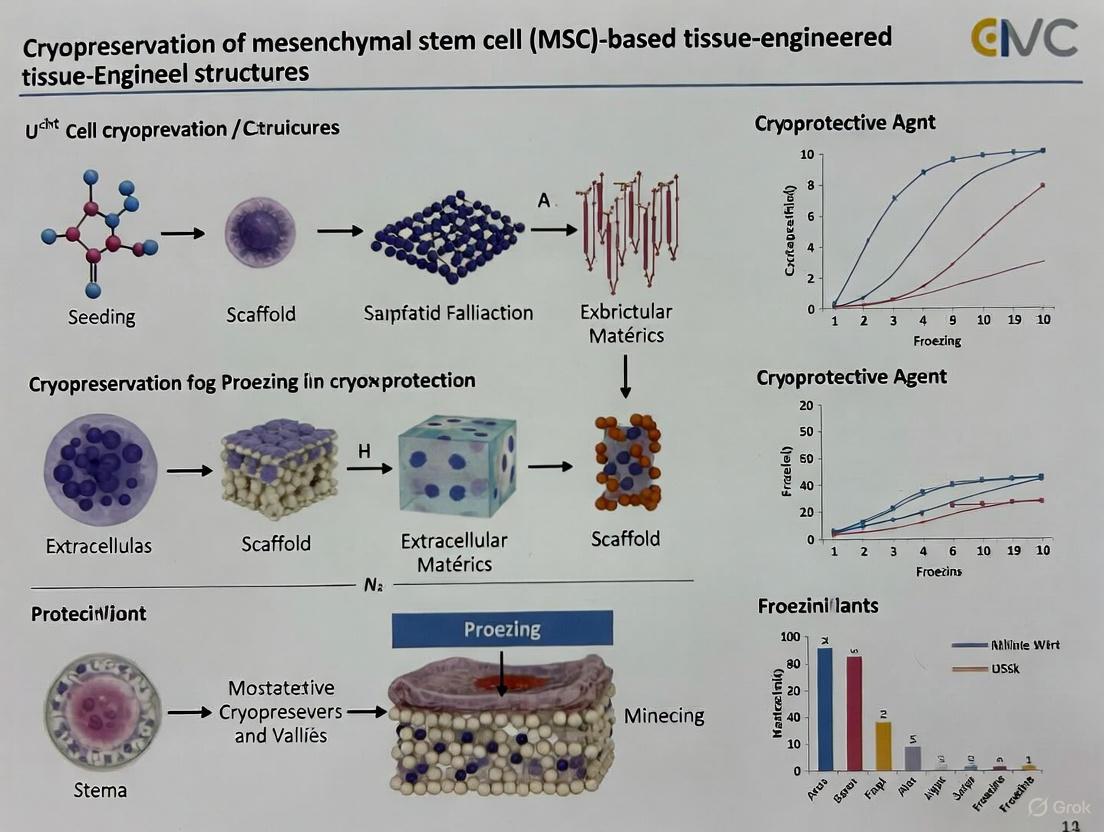
Cryopreservation Protocols for MSC-Based Structures
Maintaining MSC viability and functionality after cryopreservation is essential for tissue engineering applications. The following protocols address both cell suspensions and tissue-engineered constructs.
Cryopreservation of MSC Suspensions
Protocol: Slow Freezing Method for MSC Suspensions
- Principle: Gradual cooling minimizes intracellular ice crystal formation by allowing cellular dehydration [4] [9].
- Materials:
- Procedure:
- Harvesting: Detach MSCs using standard methods (e.g., trypsin/EDTA) and prepare a single-cell suspension.
- CPA Addition: Gently resuspend the cell pellet in pre-chilled CPA solution. Use a cell concentration of 1-5 × 10^6 cells/mL [4] [9].
- Packaging: Aliquot the cell suspension into cryogenic vials.
- Freezing: Place vials in a controlled-rate freezer. Cool at a rate of -1°C/min to -40°C, then at -5°C/min to -100°C, before transferring to liquid nitrogen vapor phase (-135°C to -150°C) for long-term storage [4] [9]. Alternatively, use a freezing container at -80°C for 24 hours before transfer.
- Thawing: Rapidly warm vials in a 37°C water bath with gentle agitation until only a small ice crystal remains.
- CPA Removal: Gently transfer thawed cell suspension to a centrifuge tube containing pre-warmed culture medium. Centrifuge at 300 × g for 5 minutes. Discard supernatant and resuspend cell pellet in fresh culture medium [4].
Cryopreservation of Tissue-Engineered Constructs
Protocol: Cryopreservation of MSC-Seeded Bioscaffolds
- Principle: Cryopreservation of 3D structures requires optimized CPA penetration to protect cells throughout the scaffold matrix [9] [15].
- Materials:
- PRP-SF Bioscaffold or other 3D construct.
- CPA: 10% DMSO or 10% DMSO + 0.2M Sucrose in appropriate medium [15].
- Cryovials suitable for 3D constructs.
- Procedure:
- Construct Preparation: Seed MSCs onto the scaffold according to established protocols and allow for sufficient cell attachment.
- CPA Equilibration: Immerse the cell-seeded construct in CPA solution. Incubate at 4°C for 30-60 minutes to allow full CPA penetration [15].
- Packaging: Transfer the construct and CPA solution to an appropriate cryogenic vial.
- Freezing: Use a controlled-rate freezer, applying a slow cooling rate of -1°C/min to -80°C, before transferring to liquid nitrogen for storage [15].
- Thawing and CPA Removal: Rapidly thaw constructs in a 37°C water bath. Immediately transfer to a large volume of pre-warmed culture medium to dilute and remove CPA. Perform multiple washes if necessary [15].
Essential Research Reagent Solutions
Table 3: Key Reagents for MSC Research and Cryopreservation
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Cryoprotective Agents (CPAs) | Dimethyl Sulfoxide (DMSO), Glycerol, Ethylene Glycol, Sucrose, Trehalose [4] [9] | Protect cells from freezing damage; DMSO penetrates cells, while sucrose/trehalose provide extracellular protection [4] [9]. |
| Culture Media | Serum-Free Medium (SFM), MSCGM-CD, DMEM-low glucose with FBS [12] | Support MSC expansion while minimizing batch-to-batch variation and safety risks associated with serum [12]. |
| Differentiation Kits | Osteogenic: Alizarin Red S; Adipogenic: Oil Red O; Chondrogenic: Alcian Blue [12] [13] [15] | Identify trilineage differentiation potential through specific histochemical staining [12]. |
| Flow Cytometry Antibodies | CD105, CD73, CD90 (positive); CD45, CD34, CD14/CD11b, CD79α/CD19, HLA-DR (negative) [5] [12] [1] | Verify MSC immunophenotype according to ISCT standards [5]. |
| Senescence Assay Kits | Senescence-associated β-galactosidase (SA-β-gal) Staining Kit [11] [10] | Detect cellular senescence in late-passage MSCs [11]. |
Experimental Workflow for MSC Characterization
The following diagram outlines a comprehensive workflow for the isolation, expansion, characterization, and cryopreservation of MSCs from different sources, highlighting key quality control checkpoints.
The selection of an optimal MSC source is application-dependent, requiring careful consideration of proliferation capacity, differentiation potential, and post-thaw functionality. Bone marrow and adipose tissue represent well-characterized sources with robust differentiation profiles, while umbilical cord and amniotic membrane offer superior proliferation capacity and lower senescence, making them particularly valuable for allogeneic banking strategies. The cryopreservation protocols detailed herein provide a foundation for maintaining MSC viability and function in both suspended and scaffold-integrated forms, supporting the development of standardized, off-the-shelf products for tissue engineering and regenerative medicine. As the field advances, further optimization of serum-free cryopreservation protocols and scaffold-specific freezing methods will be essential for clinical translation.
The Critical Need for Cryopreservation in Regenerative Medicine and Off-the-Shelf Therapies
Cryopreservation has emerged as an indispensable technological pillar in regenerative medicine and the development of "off-the-shelf" cell therapies. Mesenchymal stem cells (MSCs), with their potent immunomodulatory properties, self-renewal capacity, and multi-lineage differentiation potential, represent a cornerstone of this therapeutic revolution [4] [1]. These cells can be isolated from diverse tissues including bone marrow, adipose tissue, umbilical cord, and amniotic membrane, offering remarkable versatility for clinical applications [4]. The minimal criteria for defining MSCs—plastic adherence, specific surface marker expression (CD105, CD73, CD90), and tri-lineage differentiation potential—establish a foundation for quality assessment that must be maintained through the cryopreservation process [4] [1].
The paradigm of regenerative medicine is shifting from autologous (patient-specific) therapies toward allogeneic (donor-derived) "off-the-shelf" products that can be manufactured at scale and made readily available when needed [16]. This transition is driven by the significant limitations of autologous approaches, including high costs (often exceeding $400,000 per patient), manufacturing scalability challenges, and variable cell quality from patients who may be immunocompromised or heavily pre-treated [16]. Cryopreservation addresses these challenges by enabling long-term storage of quality-tested MSC-based products, facilitating inventory management, ensuring product stability during transportation, and allowing for comprehensive safety testing before clinical use [4] [17]. Without effective cryopreservation strategies, the vision of widely accessible, standardized, off-the-shelf regenerative therapies would remain clinically unattainable.
Critical Needs Analysis: Why Cryopreservation is Non-Negotiable
Enabling Off-the-Shelf Therapeutic Availability
The development of allogeneic MSC therapies faces a fundamental logistical challenge: how to have living cellular products immediately available for acute clinical needs while maintaining consistent quality and potency. Cryopreservation provides the only viable solution by suspending biological time, effectively pausing cellular metabolism and preventing phenotypic drift [4]. This capability is particularly crucial for tissue-engineered constructs (TECs) containing MSCs, which have short functional lifespans measured in days rather than weeks or months [18]. The ability to cryopreserve such constructs enables the creation of biobanks that can serve immediate clinical needs in scenarios such as extensive burns, acute myocardial infarction, or traumatic injuries where timely intervention is critical for patient survival [18].
Preserving MSC Functionality and Potency
Beyond mere cell survival, effective cryopreservation must maintain the critical therapeutic attributes of MSCs. These include their immunomodulatory capacity mediated through direct cell-cell contact and paracrine activity [19], differentiation potential, and secretory functions. Research demonstrates that cryopreservation helps maintain genomic stability by avoiding the epigenetic alterations and random genomic losses that can occur with continuous passaging [4]. The functional preservation of MSC immunomodulation—through interactions with T cells, B cells, natural killer cells, macrophages, and dendritic cells—must be verified post-thaw to ensure therapeutic efficacy [19].
Supporting Scalable Manufacturing and Commercialization
The transition from laboratory-scale production to commercially viable regenerative medicine products requires robust cryopreservation protocols that integrate seamlessly with Good Manufacturing Practice (GMP) standards [17]. Cryopreservation enables quality control testing, batch release validation, and flexible distribution logistics that are essential for regulatory approval and market authorization. For Advanced Therapy Medicinal Products (ATMPs), maintaining a frozen state provides the necessary stability for conducting comprehensive safety assessments, including screening for tumorigenicity and genetic instability [17]. This is particularly important for pluripotent stem cell-derived products, where residual undifferentiated cells must be rigorously quantified and controlled [17].
Quantitative Analysis of Cryopreservation Outcomes
Table 1: Comparative Analysis of MSC Cryopreservation Methods and Outcomes
| Parameter | Slow Freezing | Vitrification | Scaffold-Integrated Cryopreservation |
|---|---|---|---|
| Typical Cooling Rate | ~1°C/min to -80°C, then transfer to LN₂ [16] | Ultra-rapid cooling (>20,000°C/min) [4] | Variable: -0.5°C to -2°C/min [18] |
| CPA Concentration | Low (5-10% DMSO) [16] | High (≥40% total CPA concentration) [4] | 10% DMSO or DMSO-free alternatives [18] |
| Typical Post-Thaw Viability | 70-80% [4] | Highly variable (20-90%) [4] | ~50% minimum required for TECs [18] |
| Key Advantages | Simplicity, scalability, minimal contamination risk [4] | Avoids intracellular ice crystal formation [4] | Maintains 3D architecture and cell-matrix interactions [18] |
| Primary Limitations | CPA toxicity, osmotic stress during addition/removal [4] | Technical complexity, CPA toxicity, challenging for large volumes [4] | Uneven CPA penetration, complex optimization [18] |
| Optimal Storage Temperature | -196°C (liquid nitrogen) [4] | -196°C (liquid nitrogen) [4] | -80°C to -196°C, depending on construct [18] |
Table 2: Impact of Cryopreservation on MSC Functional Properties
| Functional Attribute | Pre-Cryopreservation Status | Post-Thaw Recovery Assessment | Key Findings from Literature |
|---|---|---|---|
| Immunomodulatory Capacity | Suppression of T-cell proliferation [19] | Co-culture with activated T-cells | Maintained if >70% viability achieved [19] |
| Paracrine Secretion | VEGF, HGF, FGF, PGE2 production [19] | ELISA/multiplex analysis of supernatant | Varies with CPA and freezing rate [19] |
| Differentiation Potential | Osteogenic, chondrogenic, adipogenic lineages [4] | Lineage-specific induction and staining | Generally preserved with optimal protocols [4] |
| Surface Marker Expression | ≥95% CD73, CD90, CD105; ≤2% hematopoietic markers [1] | Flow cytometry at P2-P3 post-thaw | Typically maintained with slow freezing [4] |
| Metabolic Activity | Normal mitochondrial function | MTT/XTT assay at 24-72h post-thaw | Transient reduction, recovery in 48-72h [18] |
Methodological Framework: Standardized Protocols for MSC Cryopreservation
Protocol 1: Slow Freezing for MSC Suspensions
Principle: Controlled-rate freezing allows gradual cellular dehydration, minimizing intracellular ice crystal formation through a combination of permeating (e.g., DMSO) and non-permeating (e.g., sucrose) cryoprotective agents (CPAs) [4].
Materials and Reagents:
- Cryogenic vials (internal thread, sterile)
- Programmable freezer or isopropanol-based freezing container
- DMSO (pharmaceutical grade)
- FBS (clinical grade if for therapeutic use) or serum-free alternatives
- Basal medium (DMEM/F12, α-MEM)
- Sucrose, trehalose, or other non-permeating CPAs
Procedure:
- Harvesting and Preparation: Harvest MSCs at 80-90% confluence using standard detachment methods. Centrifuge and resuspend in growth medium at 1-5×10^6 cells/mL.
- CPA Addition: Prepare freezing medium containing 10% DMSO, 20% FBS (if permitted for application), and 0.2M sucrose in basal medium. Gently mix cell suspension with freezing medium in 1:1 ratio to achieve final concentration of 5×10^5 to 2×10^6 cells/mL with 5% DMSO.
- Aliquoting: Dispense 1-2mL aliquots into cryogenic vials. Label with unique identifiers including cell type, passage, date, and concentration.
- Controlled Cooling: Place vials in programmable freezer with following profile:
- 4°C for 30 minutes (equilibration)
- -1°C/min to -40°C (controlled freezing)
- -5°C/min to -80°C (rapid cooling)
- Transfer to liquid nitrogen vapor phase (-150°C to -196°C) for long-term storage Alternative: Use isopropanol freezing container at -80°C for 24h, then transfer to liquid nitrogen.
- Thawing and CPA Removal: Rapidly warm vials in 37°C water bath with gentle agitation until small ice crystal remains. Immediately transfer contents to pre-warmed growth medium containing 10% FBS at 10x volume of frozen suspension. Centrifuge at 300×g for 5 minutes to remove CPA.
- Post-Thaw Assessment: Resuspend in complete growth medium, determine viability via trypan blue exclusion, and plate at 5,000-10,000 cells/cm². Monitor recovery for 24-72h before experimental use [4].
Protocol 2: Vitrification for MSC Spheroids and Tissue-Engineered Constructs
Principle: Ultra-rapid cooling achieves a glass-like state without ice crystal formation using high CPA concentrations, suitable for complex structures where controlled cooling is challenging [4].
Materials and Reagents:
- Vitrification solutions (VS1, VS2) with increasing CPA concentrations
- Open-pull straws or specialized vitrification devices
- Liquid nitrogen and storage tanks
- Sterile surgical blades or biopsy punches for construct standardization
Procedure:
- Equilibration: Transfer MSC spheroids or small tissue constructs (<1mm³) to VS1 containing 10% DMSO + 10% ethylene glycol in basal medium with 20% FBS for 10-15 minutes at room temperature.
- Vitrification Solution Exposure: Transfer to VS2 containing 20% DMSO + 20% ethylene glycol + 0.5M sucrose for 1 minute at room temperature.
- Loading and Cooling: Place 2-5 constructs on vitrification device and immediately plunge into liquid nitrogen. Ensure complete vitrification within seconds.
- Storage: Transfer to pre-cooled cryovials or sealed containers under liquid nitrogen.
- Warning and CPA Removal: Warm in 37°C water bath for 60-90 seconds. Immediately transfer to descending sucrose concentrations (1.0M, 0.5M, 0.25M, 0M) for 5 minutes each at room temperature.
- Functional Assessment: Transfer to growth medium and assess viability (FDA/PI staining), architecture (histology), and secretory function (ELISA) at 24-72h post-warming [4].
Protocol 3: Cryopreservation of MSC-Seeded Hydrogel Scaffolds
Principle: Tailored protocol addressing challenges of 3D constructs including uneven CPA penetration, differential cooling rates, and maintenance of cell-matrix interactions post-thaw [18].
Materials and Reagents:
- Hydrogel scaffolds (alginate, collagen, hyaluronic acid-based)
- DMSO or DMSO-free CPAs (e.g., glycerol, ethylene glycol)
- Cryocontainers optimized for scaffold dimensions
- CPA loading systems for enhanced penetration
Procedure:
- Pre-cryopreservation Culture: Seed MSCs onto scaffolds at optimal density (typically 1-5×10^6 cells/cm³) and culture for 3-7 days to establish ECM production and cell-matrix interactions.
- CPA Loading: Incubate constructs in CPA solution (10% DMSO + 0.2M trehalose in serum-free medium) for 60 minutes at 4°C with gentle agitation to ensure uniform penetration.
- Packaging: Place individual constructs in appropriate cryocontainers that prevent compression while allowing adequate heat transfer.
- Optimized Freezing: Use controlled-rate freezing at -0.5°C/min to -40°C, then -1°C/min to -80°C. For alginate-based hydrogels, storage at -80°C may be sufficient; for clinical applications, transfer to liquid nitrogen vapor phase.
- Thawing and CPA Removal: Rapid thaw at 37°C for 2-3 minutes, then transfer to descending sucrose solutions (0.5M, 0.25M, 0M) for 10 minutes each with gentle agitation.
- Quality Assessment: Evaluate using comprehensive criteria including:
- Cell viability (Live/Dead staining with confocal microscopy)
- Morphology and distribution (histology, SEM)
- Proliferative activity (DNA quantification, Ki67 staining)
- Secretory activity (VEGF, IL-6, IL-8 ELISA)
- Mechanical properties (if functionally relevant) [18]
Visualizing Cryopreservation Workflows and Cellular Impacts
Cryopreservation Method Selection and Workflow
Post-Thaw MSC Immunomodulatory Mechanisms
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for MSC Cryopreservation
| Reagent Category | Specific Examples | Function & Mechanism | Application Notes |
|---|---|---|---|
| Permeating CPAs | DMSO (5-10%), Glycerol (10-20%), Ethylene Glycol (10-20%) | Penetrate cell membrane, reduce ice crystal formation, depress freezing point | DMSO remains gold standard despite toxicity concerns; concentration optimization critical [4] |
| Non-Permeating CPAs | Sucrose (0.2-0.5M), Trehalose (0.2-0.4M), Hydroxyethyl starch | Create osmotic gradient, promote cellular dehydration, stabilize membranes | Particularly valuable for DMSO-free protocols; trehalose shows superior membrane stabilization [4] [18] |
| Cryopreservation Media | Commercial serum-free formulations (e.g., CryoStor, STEM-CELLBANKER) | Defined composition, regulatory compliance, batch-to-batch consistency | Essential for clinical applications; often contain DMSO + non-permeating CPAs in optimized ratios [16] |
| Scaffold Materials | Alginate, Collagen, Hyaluronic acid, Fibrin, Synthetic polymers (PLGA, PCL) | Provide 3D architecture, mimic native ECM, influence cell response | Alginate demonstrates intrinsic cryoprotective properties; architecture affects cryopreservation outcome [18] |
| Viability Assessment | FDA/PI staining, Calcein-AM/EthD-1, MTT/XTT assays, ATP quantification | Determine membrane integrity, metabolic activity, functional recovery | Multi-parameter assessment essential; viability alone insufficient for functional prediction [18] |
| Functional Assays | T-cell suppression assays, Cytokine profiling (ELISA/MSD), Differentiation kits | Verify immunomodulatory capacity, secretory function, differentiation potential | Critical for confirming therapeutic potency post-thaw; should align with intended mechanism of action [19] |
Current Challenges and Future Directions
Overcoming DMSO Toxicity and Safety Concerns
Despite its effectiveness, DMSO poses significant challenges for clinical translation of cryopreserved MSC products. At concentrations as low as 0.5-1%, DMSO demonstrates cytotoxicity in sensitive cell types like neurons and retinal ganglion cells [16]. More concerning are the clinical adverse events associated with DMSO administration, ranging from nausea and headaches to rare but severe reactions including respiratory distress and cardiovascular events [16]. The current necessity for post-thaw washing to remove DMSO introduces additional risks including contamination, cell loss through centrifugation, and procedural complexity at the point-of-care [16]. Research initiatives are actively pursuing DMSO-free cryopreservation strategies using combinations of non-toxic permeating CPAs (e.g., ethylene glycol, glycerol) with advanced non-permeating agents (trehalose, sucrose) and macromolecular additives (hydroxyethyl starch, polyvinylpyrrolidone) [4] [16].
Advancing Cryopreservation of Complex Tissue-Engineered Constructs
The transition from cryopreserving MSC suspensions to preserving complex tissue-engineered structures introduces multifaceted challenges including uneven CPA penetration, differential cooling rates throughout the construct, and maintenance of critical cell-matrix interactions post-thaw [18]. Research indicates that scaffold architecture significantly influences cryopreservation outcomes, with porous scaffolds demonstrating superior post-thaw viability compared to non-porous structures [18]. Future directions include the development of scaffold-specific cryopreservation protocols, intelligent biomaterials with inherent cryoprotective properties, and advanced warming technologies such as nanowarming that provide uniform heating throughout 3D structures [18].
Integrating Advanced Technologies and Quality-by-Design
The field is rapidly moving toward the implementation of "deep technology" solutions including artificial intelligence for predictive modeling of optimal cryopreservation parameters, automated monitoring systems for cryostorage inventory, and advanced analytics for real-time quality assessment [17] [20]. Quality-by-design principles are being applied to establish critical quality attributes (CQAs) that correlate with in vivo efficacy rather than relying solely on viability metrics [17]. These technological advancements, combined with improved understanding of MSC biology and the molecular mechanisms of freezing damage, promise to transform cryopreservation from an empirical art to a predictive science, ultimately accelerating the clinical translation of off-the-shelf MSC therapies for a broad spectrum of human diseases [4] [1] [20].
Cryopreservation is a cornerstone technology for the long-term storage of mesenchymal stem cells (MSCs), which are vital for tissue engineering and regenerative medicine applications. The therapeutic potential of MSCs relies on their functional integrity after thawing, which is directly threatened by the physical and chemical stresses encountered during freeze-thaw cycles [21] [4]. The fundamental mechanisms of cryoinjury—intracellular ice crystal formation and osmotic stress—represent the primary challenges to achieving high post-thaw viability and functionality. For MSC-based tissue-engineered structures, which often involve complex, three-dimensional architectures, these challenges are magnified, making a precise understanding of these principles essential for protocol development [22]. This application note details the underlying mechanisms, provides quantitative models for experimental design, and outlines protocols to mitigate these damaging processes.
Fundamental Mechanisms of Cryoinjury
Intracellular Ice Crystal Formation
Intracellular ice formation (IIF) is widely considered a lethal event during cryopreservation. Its formation is governed by the competition between the cooling rate and the rate of water transport across the cell membrane.
- Nucleation and Growth: When the temperature falls below the freezing point, water molecules begin to arrange into a crystalline structure. Ice typically nucleates first in the extracellular space. At slow cooling rates, intracellular water has sufficient time to exit the cell, thus avoiding IIF. At high cooling rates, however, water cannot exit the cell quickly enough, leading to supercooling and ultimately, intracellular nucleation [21] [23].
- Mechanical Damage: Intracellular ice crystals can disrupt critical intracellular structures, including microtubules, DNA, and organelles, leading directly to cell death [21].
- Recrystallization During Thawing: A significant threat occurs during the rewarming phase. As the temperature rises between -60 °C and -15 °C, existing small ice crystals can melt and refreeze into larger, more damaging crystals in a process known as recrystallization, which causes additional mechanical injury [21] [24].
Osmotic Stress and Solute Damage
The formation of extracellular ice initiates a sequence of osmotic imbalances that pose a major threat to cell survival.
- Cell Dehydration: Extracellular ice formation excludes solutes, increasing the concentration of the extracellular solution. This creates an osmotic gradient that draws water out of the cell, leading to cellular dehydration and excessive shrinkage [21] [23]. While some dehydration is beneficial to reduce IIF, excessive shrinkage can cause irreversible damage to the cell membrane and cytoskeleton.
- Solute Effect: As cells dehydrate, the concentration of intracellular electrolytes and other solutes rises dramatically. This can lead to protein denaturation, lipid peroxidation, and the disruption of crucial metabolic functions, a phenomenon collectively termed "solute damage" or "solution effects" [23].
- Oxidative Stress: The cryopreservation process itself can induce the generation of reactive oxygen species (ROS), such as superoxide radicals and hydrogen peroxide. This oxidative stress can damage lipids, proteins, and DNA, further compromising cell viability [21].
Table 1: Key Parameters and Their Impact on Mouse Oocyte Cryopreservation (Example Cell Type)
| Parameter | Impact/Recommended Value | Key Finding |
|---|---|---|
| Optimal Cooling Rate | 0.4–1.8 °C·min⁻¹ | Minimizes intracellular ice formation and dehydration damage [24]. |
| Initial DMSO Concentration | 0.1–0.3 M | Balances cryoprotection with cytotoxicity for efficient recovery [24]. |
| Recommended Warming Rate | High-power pulse | Reduces increase in intracellular ice volume during recrystallization phase [24]. |
| Safe Cryostorage Temperature | < -160 °C | Prevents recrystallization during storage and handling [24]. |
Experimental Protocols for Investigating Cryoinjury
Protocol: Modeling Transmembrane Transport and Intracellular Crystallization
This protocol is adapted from a computational study on mouse oocytes and provides a framework for modeling cryoinjury in cells [24].
1. Objective To predict trends in intracellular water content, cryoprotectant (CPA) concentration, and ice crystal volume during a freeze-thaw cycle using a cell-scale numerical model.
2. Materials
- A validated numerical model incorporating coupled water/CPA transport and ice nucleation/growth.
- Cell-specific parameters (e.g., membrane permeability to water and CPA, surface area, initial volume).
- Temperature profile data for the planned freeze-thaw cycle.
- Ternary solution data (e.g., water, NaCl, DMSO).
3. Methodology
- Model Input: Define the temperature profile of the entire freeze-thaw process as the input condition.
- Parameter Calculation: The model calculates two primary sets of parameters simultaneously:
- Transmembrane Transport: The flux of water and CPA (e.g., DMSO) across the cell membrane in non-ideal, non-dilute solutions.
- Nucleation and Growth: The nucleation temperature and subsequent growth of intracellular ice crystals during cooling.
- Recrystallization: The growth of existing ice crystals during the rewarming phase.
- Output Analysis: The model outputs the temporal evolution of intracellular CPA concentration, free water content, and the volume fraction of intracellular ice.
4. Key Applications
- Determine the optimal cooling and warming rates for a specific cell type.
- Identify the CPA concentration that minimizes both intracellular ice formation and osmotic stress.
- Simulate the damaging impact of cryopreservation vial handling (e.g., "pick-and-place" operations) by modeling temperature fluctuations that induce recrystallization.
Protocol: Slow Freezing of Mesenchymal Stem Cells
This is a standard operational protocol for the cryopreservation of MSCs using the slow freezing method [4].
1. Objective To preserve MSCs for long-term storage using a controlled slow freezing process to minimize intracellular ice formation.
2. Materials
- Culture medium and trypsin/EDTA for cell detachment.
- Cryopreservation medium: Culture medium supplemented with a penetrating CPA (e.g., 10% DMSO) and potentially a non-penetrating CPA (e.g., sucrose or trehalose).
- Fetal bovine serum (FBS) or human platelet lysate (hPL) as a protein source.
- Controlled-rate freezer or isopropanol-based freezing container.
- Cryogenic vials.
- Liquid nitrogen storage tank.
3. Methodology
- Cell Harvesting: Harvest MSCs at the desired passage using standard trypsinization. Centrifuge and resuspend the cell pellet in cold cryopreservation medium at a typical concentration of 1 x 10^6 to 1 x 10^7 cells/mL.
- Packaging: Aliquot the cell suspension into cryogenic vials.
- Initial Cooling: Place the vials at 4°C for 30-60 minutes for temperature equilibration.
- Controlled Freezing:
- Option A (Controlled-Rate Freezer): Place vials in the freezer and initiate a program that cools at a rate of -1°C/min to -40°C, then at -5°C/min to -100°C, before transferring to liquid nitrogen.
- Option B (Freezing Container): Place vials in a pre-cooled isopropanol freezing container and transfer directly to a -80°C freezer for 24 hours. The container provides an approximate cooling rate of -1°C/min.
- Long-term Storage: After 24 hours, promptly transfer the vials to the vapor or liquid phase of a liquid nitrogen storage tank (-150°C to -196°C).
- Thawing: Rapidly thaw the vial by gentle agitation in a 37°C water bath until only a small ice crystal remains (~60-90 seconds). Immediately dilute the cell suspension drop-wise with pre-warmed culture medium and centrifuge to remove the CPA. Resuspend the cell pellet in fresh culture medium for subsequent use.
Table 2: The Scientist's Toolkit: Key Reagents for Cryopreservation Research
| Research Reagent / Material | Function / Explanation |
|---|---|
| Dimethyl Sulfoxide (DMSO) | A penetrating cryoprotectant; forms hydrogen bonds with water, depressing the freezing point and reducing ice crystal formation [21] [4]. |
| Glycerol | A penetrating cryoprotectant; less toxic than DMSO for some cell types but may be less effective [4] [23]. |
| Sucrose/Trehalose | Non-penetrating cryoprotectants; modulate extracellular osmotic pressure, aiding in controlled dehydration and reducing CPA toxicity [4]. |
| Alginate Hydrogels | Natural polymer for cell encapsulation; provides a physical barrier against ice crystals and helps regulate osmotic balance [22]. |
| Polyvinyl Alcohol (PVA) Hydrogels | Synthetic polymer for encapsulation; exhibits strong resistance to mechanical stress during freezing and reduces ice crystal formation [22]. |
| Human Platelet Lysate (hPL) | A GMP-compliant, serum-free growth supplement for MSC culture and cryopreservation media; reduces reliance on fetal bovine serum [25]. |
| Antioxidants (e.g., SOD, Catalase) | Mitigate oxidative stress by scavenging reactive oxygen species (ROS) generated during the freeze-thaw process [21]. |
Visualizing Cryoinjury Pathways and Experimental Workflows
Cryoinjury Mechanisms During Freeze-Thaw Cycle
Integrated Strategy for MSC Cryopreservation
The successful cryopreservation of MSC-based tissue-engineered structures hinges on a deliberate and balanced approach to mitigating the twin pillars of cryoinjury: intracellular ice formation and osmotic stress. As detailed in this note, this involves not only the careful selection and application of CPAs but also the precise control of thermal profiles and the emerging use of advanced biomaterials like encapsulation hydrogels. Future progress will likely depend on multidisciplinary strategies that integrate insights from cryobiology with innovations in materials science, nanotechnology, and computational modeling to achieve high viability and functionality in complex, clinically relevant tissue constructs [21] [22].
The cryopreservation of Mesenchymal Stem Cells (MSCs) is a critical step in the supply chain for both basic research and clinical cell-based therapies, enabling storage, quality control, and logistical coordination [26]. For a broader thesis on MSC-based tissue-engineered structures, understanding this impact is foundational, as the functionality of the cellular component directly influences the final product's therapeutic efficacy. The process, however, induces various stresses that can compromise core MSC functionalities, including their immunomodulatory capacity, self-renewal potential, and ability to differentiate into multiple lineages [27] [28]. This Application Note synthesizes current research to summarize these impacts and provides detailed, executable protocols for assessing MSC potency post-cryopreservation, serving as a essential resource for researchers and therapy developers in the field.
The following table synthesizes key quantitative findings from recent studies on how cryopreservation affects fundamental MSC properties.
Table 1: Impact of Cryopreservation on Core MSC Functionalities: Key Experimental Findings
| Core Functionality | Key Findings | Experimental Model | Reference |
|---|---|---|---|
| Immunomodulation | Thawed MSCs significantly arrested T-cell proliferation, but potency was significantly higher after a 24-hour acclimation period. IFN-γ secretion was also significantly diminished in freshly thawed cells. | Human Bone Marrow-derived MSCs, T-cell proliferation assay | [28] |
| Self-Renewal | Colony-forming capacity was decreased in freshly thawed MSCs. A 24-hour post-thaw acclimation period enabled recovery of this function. | Human Bone Marrow-derived MSCs, CFU-f Assay | [28] |
| Multi-Lineage Potential | MSC proliferation and multilineage differentiation were preserved after freezing Bone Marrow Aspirate Concentrate (BMAC) at -80°C for 4 weeks. In vivo, frozen BMAC improved cartilage repair equivalently to fresh BMAC in an OA rat model. | Human BMAC, in vitro differentiation & in vivo OA rat model | [29] |
| Cell Survival & Phenotype | Average post-thaw viability decreased by 11.4% with a novel DMSO-free solution (SGI) and 4.5% with DMSO-based solutions. Viable cell recovery was better with the SGI solution (92.9% vs lower for DMSO). Phenotype (CD73, CD90, CD105) was comparable across methods. | International multicenter study on Bone Marrow and Adipose-derived MSCs | [26] |
| Viability & Recovery | Post-thaw, apoptosis was significantly increased in freshly thawed cells. A 24-hour acclimation period significantly reduced apoptosis and reactivated key regenerative and angiogenic genes. | Human Bone Marrow-derived MSCs, Annexin V/PI apoptosis assay & gene expression | [28] |
Experimental Protocols for Assessing MSC Potency Post-Cryopreservation
Protocol: Colony-Forming Unit Fibroblast (CFU-f) Assay for Self-Renewal
Principle: This assay measures the clonogenic potential of MSCs, a direct indicator of their self-renewal capacity, by quantifying their ability to form discrete cell colonies from a single progenitor [29] [28].
Materials:
- Mononuclear cell fraction from fresh or thawed BMAC/MSCs [29]
- Growth Medium: αMEM supplemented with 20% FBS, 1% Penicillin/Streptomycin, and 10 ng/mL FGF-2 [29]
- 4% Paraformaldehyde (PFA)
- 1% Crystal Violet solution
Method:
- Cell Seeding: Plate mononuclear cells at a density of 300,000 cells/well in a 6-well tissue culture plate. Use a minimum of three technical replicates per condition [29].
- Culture: Cultivate cells for 14 days in Growth Medium at 37°C and 5% CO₂. Do not disturb the plates excessively to allow for colony formation.
- Fixation and Staining: On day 14, carefully aspirate the media and wash wells with PBS. Fix cells with 4% PFA for 15 minutes at room temperature. Remove PFA and stain with 1% Crystal Violet solution for 30 minutes.
- Washing and Analysis: Rinse plates gently with distilled water to remove excess stain and air-dry. Manually count colonies using a pre-defined criterion (e.g., clusters of >100 cells). A distinct clonal center must be evident for a cluster to be counted as a single colony [29].
Protocol: In Vitro Trilineage Differentiation for Multi-Lineage Potential
Principle: This functional assay confirms MSC multipotency by inducing differentiation into osteocytes, chondrocytes, and adipocytes, followed by lineage-specific staining [1] [28].
Materials:
- MSCs (Fresh, Freshly Thawed, or Acclimated)
- Commercial Trilineage Differentiation Media Kits (e.g., StemPro, Thermo Fisher Scientific)
- Fixatives and stains: Alizarin Red S (osteogenesis), Alcian Blue (chondrogenesis), Oil Red O (adipogenesis)
Method:
- Osteogenic Differentiation:
- Seed MSCs in chamber slides or well plates at a standard density.
- Culture in osteogenic differentiation media for 21 days, replacing the media twice weekly.
- After 21 days, fix cells and stain with Alizarin Red to detect calcium deposits [28].
- Chondrogenic Differentiation:
- Create a micromass by seeding a 5μL droplet of high-density cell solution (e.g., 1.6 x 10⁷ cells/mL) in the center of a well.
- Allow the micromass to adhere for 2 hours before carefully adding chondrogenic differentiation media.
- Culture for 14 days, changing media every other day.
- Fix the micromass and stain with Alcian Blue to visualize sulfated proteoglycans [28].
- Adipogenic Differentiation:
- Seed MSCs and culture until confluent.
- Switch to adipogenic induction media, followed by maintenance media as per kit instructions, for 14-21 days.
- Fix cells and stain with Oil Red O to label lipid vacuoles.
Protocol: T-Cell Proliferation Assay for Immunomodulation
Principle: This assay evaluates the functional immunomodulatory capacity of MSCs by measuring their ability to suppress the proliferation of activated immune cells [28].
Materials:
- MSCs (test groups: FC, FT, TT)
- Peripheral Blood Mononuclear Cells (PBMCs) from a healthy donor
- T-cell mitogen (e.g., Phytohemagglutinin - PHA)
- Cell proliferation dye (e.g., CFSE) or ³H-thymidine
- Flow cytometer or scintillation counter
Method:
- MSC Preparation: Seed and allow MSCs from different treatment groups (Fresh, Freshly Thawed, Thawed+24h) to adhere in a co-culture compatible plate.
- PBMC Activation: Isolate PBMCs and label with CFSE according to manufacturer's instructions. Activate T-cells within the PBMC population by adding PHA.
- Co-culture: Add activated, CFSE-labeled PBMCs directly to the MSC cultures. Establish controls containing only PBMCs with PHA (maximum proliferation) and without PHA (background proliferation).
- Analysis: After 3-5 days of co-culture, harvest non-adherent cells and analyze by flow cytometry to measure CFSE dilution in T-cells. Alternatively, measure proliferation by ³H-thymidine incorporation. The degree of suppression is calculated by comparing proliferation in co-culture wells to the maximum proliferation control [28].
Signaling Pathways and Experimental Workflows
MSC Post-Thaw Recovery Pathway
The following diagram illustrates the key cellular processes and molecular changes that occur in MSCs during the critical post-thaw acclimation period, leading to the recovery of functional potency.
Cryopreservation Experimental Workflow
This workflow outlines the key stages in a standardized protocol for evaluating the impact of cryopreservation on MSCs, from cell preparation to functional assessment.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for MSC Cryopreservation Research
| Reagent/Material | Function/Application | Examples & Notes |
|---|---|---|
| Cryoprotective Agents (CPAs) | Protect cells from ice crystal formation and osmotic damage during freeze-thaw. | Penetrating (DMSO, Glycerol): Standard but can be cytotoxic [28] [30]. Non-penetrating (Sucrose, Trehalose): Often used in DMSO-free cocktails (e.g., SGI solution: Sucrose, Glycerol, Isoleucine) [26]. |
| Culture Media for Recovery | Supports cell metabolism and repair during post-thaw acclimation. | Basal Media (αMEM, DMEM) supplemented with FBS (10-20%) and FGF-2 (10 ng/mL) [29] [28]. |
| Phenotypic Markers | Confirms MSC identity and surface marker integrity post-thaw via flow cytometry. | Positive Panel: CD73, CD90, CD105 (≥95% expression) [1] [26]. Negative Panel: CD45, CD34, CD11b, CD19, HLA-DR (≤2% expression) [1]. |
| Trilineage Differentiation Kits | Standardized systems for assessing multipotency post-thaw. | Commercial kits (e.g., StemPro, Thermo Fisher) provide optimized induction media for osteogenic, chondrogenic, and adipogenic differentiation [28]. |
| Controlled-Rate Freezer | Ensures reproducible, optimal cooling rate to minimize cryoinjury. | Standard slow freezing rate is -1°C/min to -80°C before transfer to liquid nitrogen [28]. Passive freezing containers (e.g., "Mr. Frosty") can be a lower-cost alternative [29]. |
Slow Freezing vs. Vitrification: Protocols for Preserving MSC Structures
The cryopreservation of mesenchymal stem cell (MSC)-based tissue-engineered structures is a critical enabling technology for regenerative medicine, allowing for the creation of "off-the-shelf" cellular products for therapeutic applications [31] [30]. Among available techniques, slow freezing remains the predominant and most recommended method for the cryostorage of MSCs and simple tissue constructs in both clinical and laboratory settings due to its operational simplicity, minimal contamination risk, and proven effectiveness [32] [4]. This protocol details the application of slow-freezing methodology for MSC-based tissue-engineered structures, framing it within the broader context of ensuring the viability, functionality, and clinical availability of these advanced therapeutic products.
The fundamental principle of slow freezing is the controlled, gradual dehydration of cells. By carefully managing the cooling rate, water exits the cell before freezing, minimizing the lethal formation of intracellular ice crystals [32] [4]. This process, supported by cryoprotective agents (CPAs), allows cells to enter a state of suspended animation, permitting their long-term storage in liquid nitrogen (LN2) at -196°C [33] [30].
Core Principles and Key Data
Successful slow freezing hinges on the interplay of three core components: controlled cooling, the use of CPAs, and cellular dehydration. The quantitative optimization of these parameters is summarized in the table below.
Table 1: Key Optimized Parameters for Slow Freezing of MSCs and MSC-Based Structures
| Parameter | Recommended Setting | Rationale & Impact | Key References |
|---|---|---|---|
| Cooling Rate | ~1°C/min to -80°C | Balances cellular dehydration against intracellular ice formation; too slow causes osmotic stress, too fast causes intracellular ice. | [34] [32] |
| Final Storage Temp | -196°C (Liquid Nitrogen) | Halts all metabolic and biochemical processes for long-term storage. | [4] [33] |
| Typical Post-Thaw Viability (Cell Suspensions) | 70-80% | Viability benchmark for MSC suspensions using standard slow-freezing protocols. | [32] [4] |
| Common Permeating CPA (DMSO) | 10% (v/v) | Increases membrane porosity, depresses freezing point, enables vitrification; higher concentrations become toxic. | [34] [30] |
| DMSO Equilibration Temperature | 0-4°C | Reduces the cytotoxic effects of the CPA. | [34] |
| Non-Permeating CPA (Trehalose/Sucrose) | 0.1-0.5 M | Provides extracellular stabilization, osmotic buffering, and reduces required DMSO concentration. | [34] [30] |
The choice of CPA is critical. CPAs are categorized as either permeating (e.g., Dimethyl Sulfoxide (DMSO), glycerol) or non-permeating (e.g., sucrose, trehalose), each with distinct functions and toxicities [30]. The following table compares common agents.
Table 2: Comparison of Common Cryoprotective Agents (CPAs)
| Cryoprotective Agent | Type | Mechanism of Action | Reported Toxicity | Notes on Clinical Use | |
|---|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating | Depresses freezing point, penetrates cell membrane, inhibits intracellular ice. | Moderate to High | Gold standard but can trigger allergic reactions in patients; requires thorough post-thaw washing. | [32] [4] |
| Glycerol (GLY) | Permeating | Similar to DMSO. | Lower | Lower toxicity but often results in worse cryopreservation effect compared to DMSO. | [4] |
| Ethylene Glycol (EG) | Permeating | Similar to DMSO. | Lower than DMSO | Effective in molar combinations with DMSO to reduce overall toxicity. | [34] [4] |
| Trehalose | Non-Permeating | Stabilizes membranes, inhibits ice recrystallization. | Very Low | Naturally produced by some organisms; often used in combination with permeating CPAs. | [34] [30] |
| Sucrose | Non-Permeating | Osmotic buffer, reduces osmotic shock during CPA addition/removal. | Very Low | Commonly used in CPA cocktails and as a component in thawing solutions. | [32] [4] |
| Polyvinyl Alcohol (PVA) | Non-Permeating | Ice recrystallization inhibition (IRI). | Low | A synthetic polymer that shows promise in improving cryopreservation outcomes. | [31] [35] |
Experimental Protocol: Slow Freezing of MSC Spheroids
This protocol provides a detailed methodology for the slow freezing of MSC spheroids, a fundamental tissue-engineered structure.
Materials and Reagents
- MSC Spheroids (e.g., ~500 μm diameter)
- Basal Freezing Medium: Standard cell culture medium (e.g., DMEM) supplemented with 10-20% (v/v) Fetal Bovine Serum (FBS).
- Cryoprotectant Stock Solution: 10% (v/v) DMSO in basal freezing medium.
- Complete CPA Cocktail: Basal freezing medium supplemented with 10% DMSO and optionally 0.2 M sucrose or trehalose.
- Pre-incubation Solution: "Material A" (as referenced in screening studies) or a solution containing antioxidants and glucose in culture medium [35].
- Equipment: Programmable controlled-rate freezer, cryogenic vials, -80°C mechanical freezer, liquid nitrogen storage tank, 37°C water bath or dry-thawing device, centrifuge.
Step-by-Step Procedure
Step 1: Pre-cryopreservation Treatment (Pre-incubation)
- Purpose: To enhance CPA penetration and spheroid robustness.
- Procedure: Pre-culture MSC spheroids for a few hours in a medium containing compounds that promote penetration of cryoprotective additives, such as the experimentally identified "Material A" [35]. Alternatively, pre-incubation with antioxidants and high glucose can be used to improve post-thaw viability and function [34].
- Critical Parameter: Pre-incubation time and compound concentration must be optimized for specific spheroid size and type.
Step 2: CPA Addition and Equilibration
- Purpose: To allow sufficient CPA penetration while minimizing toxicity.
- Procedure:
- Gently transfer pre-incubated spheroids to the Complete CPA Cocktail at 4°C.
- Incubate at 4°C for 15-20 minutes to allow for CPA equilibration. This cold temperature reduces CPA toxicity [34].
- Critical Parameter: Do not exceed equilibration time; prolonged exposure to high DMSO concentrations is cytotoxic.
Step 3: Controlled-Rate Freezing
- Purpose: To induce controlled cellular dehydration.
- Procedure:
- Aliquot spheroids in CPA cocktail into cryogenic vials.
- Place vials in a controlled-rate freezer.
- Initiate cooling program:
- Alternatively, if a controlled-rate freezer is unavailable, use a "step-down" method: place vials at -20°C for 24 hours, then transfer to -80°C [32].
- Critical Parameter: The cooling rate of ~1°C/min is crucial for preventing intracellular ice formation.
Step 4: Long-Term Storage
- Purpose: To maintain cells in a metabolically inactive state.
- Procedure: After reaching -80°C, promptly transfer cryovials to the vapor phase of liquid nitrogen (-150°C to -196°C) for long-term storage [32] [4].
- Critical Parameter: Avoid temperature fluctuations during storage.
Step 5: Thawing and CPA Removal
- Purpose: To rapidly reheat samples and remove toxic CPAs.
- Procedure:
- Quickly retrieve vial from LN2 and immediately place it in a 37°C water bath with gentle agitation until only a small ice crystal remains (~2-3 minutes). Using a dry-thawing device is preferred to avoid microbial contamination from water baths [4].
- Decontaminate the vial with 70% ethanol.
- Gently transfer the thawed suspension to a tube containing pre-warmed basal medium (without CPAs).
- Centrifuge at a low speed (e.g., 300 x g for 5 minutes) to pellet spheroids/cells.
- Carefully aspirate the supernatant containing the CPA.
- Resuspend the pellet in fresh culture medium.
- Critical Parameter: Thaw rapidly. Dilute CPA gently to avoid osmotic shock; a stepwise dilution may be beneficial.
Workflow and Mechanism Visualization
The following diagram illustrates the logical workflow and the cellular mechanisms of the slow-freezing protocol.
Diagram 1: Experimental Workflow for MSC Spheroid Cryopreservation.
The core cellular process during slow freezing is controlled dehydration, which is visualized in the following diagram.
Diagram 2: Cellular Dehydration Mechanism in Slow Freezing.
The Scientist's Toolkit: Essential Research Reagents
This section details key reagents and materials essential for implementing the slow-freezing protocol for MSC-based structures.
Table 3: Essential Research Reagents and Materials
| Item | Function/Description | Example Use Case |
|---|---|---|
| Programmable Controlled-Rate Freezer | Ensures precise, reproducible cooling at ~1°C/min, which is critical for success. | Standardized freezing of research and clinical-grade samples. |
| DMSO (Cell Culture Grade) | Permeating CPA; depresses freezing point and prevents intracellular ice. | Used at 10% (v/v) in freezing medium as the primary CPA. |
| Trehalose (Cell Culture Grade) | Non-permeating CPA; provides extracellular stabilization and osmotic buffering. | Added (0.1-0.5 M) to CPA cocktail to reduce DMSO toxicity and improve viability. |
| "Material A" (Penetration Enhancer) | A material identified via screening to promote CPA penetration into cell aggregates. | Pre-culture of MSC spheroids to enhance CPA distribution and increase post-thaw viability and activity [35]. |
| Liquid Nitrogen Storage System | Provides long-term storage at -196°C, halting all metabolic activity. | Secure, organized biobanking of cryopreserved constructs. |
| Dry-Thawing Device | Heats cryovials to 37°C without a water bath, minimizing contamination risk. | Safer and more GMP-compliant thawing of clinical-grade products [4]. |
| Viability/Cytotoxicity Assay Kit | Measures post-thaw cell survival (e.g., calcein-AM) and death (e.g., propidium iodide). | Standard quality control post-thaw. |
| LDH Release Assay Kit | Quantifies lactate dehydrogenase enzyme released upon cell lysis, indicating cytotoxicity. | Assessing cryoinjury, especially in high-density spheroid cultures [35]. |
| Zwitterionic CPA (e.g., OE2imC3C) | An emerging cell-impermeable cryoprotectant that increases extracellular osmolarity. | Used in combination with DMSO (e.g., 10 wt% Zwitterion, 15 wt% DMSO) for improved spheroid recovery and function [36]. |
Vitrification has emerged as a pivotal cryopreservation technique for mesenchymal stem cell (MSC)-based tissue-engineered structures, enabling their long-term preservation for regenerative medicine and drug development applications. This process transforms the aqueous cellular environment into a stable, glassy state without forming destructive ice crystals, thereby maintaining structural integrity and biological functionality post-thaw [4] [37]. For MSC-based constructs, which often comprise complex three-dimensional architectures, vitrification offers significant advantages over conventional slow-freezing methods by mitigating ice-induced damage that compromises tissue viability and function [30] [38].
The fundamental principle of vitrification involves achieving an amorphous glassy solidification through extreme elevation in viscosity, bypassing crystalline ice formation entirely [38]. This transition occurs when solutions reach sufficient viscosity (approximately 10¹³ poise) to maintain a disordered molecular arrangement characteristic of liquids while possessing the mechanical properties of solids [38]. Successful vitrification depends on navigating critical temperature transitions: the melting temperature (Tm), where freezing begins; the homogeneous nucleation temperature (Th), where ice nucleation becomes probable; and the glass transition temperature (Tg), where the solution vitrifies [37] [39]. The temperature region between Tm and Tg represents the "Dangerous Temperature Zone" (DTZ), where intracellular ice formation predominantly causes cellular damage [39].
Two principal methodologies have been developed to achieve vitrification: equilibrium and non-equilibrium approaches. These strategies balance the interplay between cryoprotectant agent (CPA) concentration, cooling rate, and sample volume to optimize vitrification outcomes for complex biological systems like MSC-based tissue constructs [4] [37].
Theoretical Foundations of Vitrification
Physical Chemistry of Glass Transitions
The vitrification process is governed by the competing dynamics of ice nucleation kinetics and water molecule diffusion limitations during cooling. When aqueous solutions are cooled below their melting temperature, they enter a supercooled liquid state where water molecules stochastically form clusters that may develop into critical ice nuclei [37]. According to classical nucleation theory, once these nuclei reach a critical size, they rapidly propagate into crystalline ice structures [37]. Vitrification prevents this transition by implementing cooling rates sufficiently rapid to avoid ice nucleation, or by using CPAs that increase solution viscosity to immobilize water molecules before they can reorganize into ice crystals [37].
The glass transition temperature (Tg) represents a critical thermodynamic parameter in vitrification protocols. Recent research demonstrates that Tg significantly influences thermal stress development during vitrification, with higher Tg values correlating with reduced cracking in vitrified solutions [40]. This relationship stems from the inverse correlation between Tg and thermal expansion coefficient – solutions with higher Tg exhibit lower thermal expansion, thereby generating less stress during temperature cycling [40]. This insight is particularly relevant for scaling vitrification protocols to larger MSC-based tissue constructs, where thermal stress management remains a primary challenge.
Comparative Principles: Equilibrium vs. Non-Equilibrium Vitrification
The two vitrification approaches employ distinct physical mechanisms to achieve the glassy state. Equilibrium vitrification emphasizes controlled osmotic balance between cells and their extracellular environment during CPA introduction [4]. This method utilizes precisely managed CPA concentration gradients and exposure times to enable gradual cellular dehydration and CPA permeation before rapid cooling. The sequential equilibration minimizes osmotic shock and volume stress, which is particularly beneficial for sensitive MSC-based constructs with complex geometries [4].
In contrast, non-equilibrium vitrification prioritizes ultra-rapid cooling kinetics combined with high CPA concentrations to achieve vitrification [4]. This approach leverages extremely high cooling rates (potentially exceeding 10⁵ °C/min) to traverse the dangerous temperature zone before ice nucleation can initiate [41] [39]. While requiring higher CPA concentrations, the dramatically increased cooling rates significantly reduce the probability of intracellular ice formation, making this method suitable for vitrifying smaller MSC constructs or cellular suspensions where ultra-rapid heat transfer is achievable.
Figure 1: Workflow comparison of equilibrium versus non-equilibrium vitrification approaches, highlighting distinct pathways to achieving the glassy state.
Comparative Analysis of Vitrification Approaches
Technical Parameter Comparison
The selection between equilibrium and non-equilibrium vitrification strategies involves balancing multiple technical parameters with specific requirements of MSC-based tissue constructs. The table below summarizes the key distinguishing characteristics of each approach:
Table 1: Comparative technical parameters of equilibrium versus non-equilibrium vitrification approaches
| Parameter | Equilibrium Vitrification | Non-Equilibrium Vitrification |
|---|---|---|
| CPA Concentration | Moderate (4-6 M) [4] | High (6-8 M) [4] [42] |
| Cooling Rate | Moderate (100-10,000°C/min) [4] | Ultra-rapid (>100,000°C/min) [41] [39] |
| CPA Exposure Time | Extended (10-15 minutes) [4] | Brief (seconds to <1 minute) [4] |
| Osmotic Stress | Controlled through gradual equilibration [4] | High due to rapid CPA addition [42] |
| CPA Toxicity Risk | Moderate [42] | Elevated [42] |
| Optimal Sample Size | Larger constructs (>1 mm³) [30] | Small volumes (<50 μL) [41] |
| Thermal Stress | Moderate [38] | Lower during cooling, higher during warming [38] |
| Technical Complexity | Moderate [4] | High [4] |
Advantages and Limitations for MSC-Based Constructs
For MSC-based tissue-engineered structures, each vitrification approach presents distinct advantages and limitations. Equilibrium vitrification offers superior compatibility with larger, three-dimensional constructs by minimizing osmotic shock through controlled CPA permeation [4] [30]. The gradual dehydration process helps maintain cell-cell and cell-matrix interactions critical for post-thaw functionality in tissue-engineered products. However, this method requires precise optimization of CPA addition/removal kinetics and carries increased processing time, which may impact workflow efficiency in clinical settings [4].
Non-equilibrium vitrification provides exceptional protection against intracellular ice formation by achieving remarkably high cooling rates, making it ideal for preserving MSC suspensions or small tissue spheroids [41] [39]. The dramatically reduced processing time minimizes biochemical alterations during the cryopreservation workflow. Nevertheless, this approach necessitates high CPA concentrations that elevate toxicity risks and imposes strict sample volume limitations due to heat transfer constraints [4] [42]. The implementation often requires specialized equipment such as nylon membrane carriers [41] or microdroplet generators [39] to achieve the necessary ultra-rapid cooling rates.
Experimental Protocols for Vitrification of MSC Constructs
Equilibrium Vitrification Protocol for MSC Spheroids
This protocol describes the stepwise procedure for vitrifying MSC-based spheroids or small tissue constructs using the equilibrium approach, optimized for preserving viability and functionality.
Materials and Reagents:
- MSC spheroids (100-200 μm diameter)
- Base medium (e.g., DMEM-LG)
- Permeating CPAs: DMSO, ethylene glycol (EG)
- Non-permeating CPAs: sucrose, trehalose
- Fetal bovine serum (FBS)
- Vitrification solution: 2.5-3.0 M DMSO, 2.5-3.0 M EG, 0.5 M sucrose in base medium with 20% FBS
- Equilibration solution: 1.5 M DMSO, 1.5 M EG in base medium with 20% FBS
- Dilution solutions: Sucrose solutions (0.8 M, 0.6 M, 0.4 M, 0.2 M) in base medium with 20% FBS
- Cryovials or specialized vitrification devices
- Liquid nitrogen storage system
Procedure:
- Pre-cooling Preparation: Maintain all solutions and equipment at 4°C throughout the procedure to minimize CPA toxicity.
- Equilibration Phase: Transfer MSC spheroids to equilibration solution for 5-7 minutes at 4°C. This gradual exposure minimizes osmotic shock while initiating cellular dehydration.
- Vitrification Solution Exposure: Briefly transfer spheroids (<1 minute) to vitrification solution at 4°C. Monitor for proper shrinkage indicating sufficient dehydration.
- Loading and Sealing: Quickly place 2-5 spheroids in minimal vitrification solution volume into cryovials or specialized vitrification devices.
- Plunge Cooling: Immediately immerse samples directly into liquid nitrogen, ensuring complete submersion.
- Storage: Transfer to long-term liquid nitrogen storage at -196°C.
- Warning Procedure: Rapidly warm samples by immersing in 37°C water bath with gentle agitation until completely thawed.
- Stepwise CPA Removal:
- Transfer spheroids to 0.8 M sucrose solution for 3 minutes
- Sequentially transfer through 0.6 M, 0.4 M, and 0.2 M sucrose solutions (3 minutes each)
- Finally, transfer to base medium for washing before functional assessment
Quality Control: Post-thaw viability should exceed 80% as assessed by membrane integrity staining. Functional assessment should include MSC differentiation potential, immunomodulatory capacity, and metabolic activity [4] [30].
Non-Equilibrium Vitrification Protocol Using Ultra-Rapid Cooling
This protocol implements the non-equilibrium approach for MSC suspensions or small aggregates using ultra-rapid cooling technology for maximal ice formation inhibition.
Materials and Reagents:
- MSC suspension (1×10⁶ cells/mL) or small aggregates (<100 μm)
- Base medium (e.g., DMEM-LG)
- CPA cocktail: 6.0 M DMSO + 0.5 M trehalose in base medium or 6.0 M EG + 0.5 M trehalose in base medium
- Nylon membrane strips (20×20 mm) or droplet vitrification system
- FBS
- Liquid nitrogen
- Forceps and handling tools
Procedure:
- CPA Loading: Suspend MSC samples in CPA cocktail at room temperature for precisely 30-60 seconds.
- Sample Loading:
- For membrane approach: Pipette 50 μL MSC suspension onto nylon membrane strip
- For droplet approach: Generate <10 μL droplets containing MSC suspension directly into liquid nitrogen using specialized apparatus
- Evaporation: Allow brief evaporation (30 seconds) under laminar flow to minimize residual solution volume [41].
- Plunge Cooling: Rapidly immerse membrane strip or droplets into liquid nitrogen, achieving cooling rates >100,000°C/min [41].
- Storage: Transfer to sealed containers for long-term storage in liquid nitrogen.
- Warning: Rapidly warm samples by transferring to pre-warmed (37°C) base medium with gentle agitation.
- CPA Removal:
- Directly transfer to culture medium with 10% FBS
- Centrifuge at 300×g for 5 minutes to remove CPAs
- Resuspend in fresh culture medium for assessment
Quality Control: Assess cell integrity (>85%) and viability (>65%) post-thaw. Evaluate surface marker expression (CD105, CD73, CD90) and apoptosis markers to ensure maintained MSC identity and minimal cryo-damage [41].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of vitrification protocols for MSC-based constructs requires specific reagents and specialized equipment. The following table catalogues essential research tools and their functional applications in vitrification workflows:
Table 2: Essential research reagents and materials for vitrification protocols
| Category | Specific Examples | Function/Application |
|---|---|---|
| Permeating CPAs | DMSO, ethylene glycol, glycerol, propylene glycol [30] [42] | Penetrate cell membranes, depress freezing point, inhibit intracellular ice formation [30] |
| Non-Permeating CPAs | Sucrose, trehalose, ficoll, polyvinyl pyrrolidone [30] | Extracellular ice inhibition, osmotic buffering during CPA addition/removal [30] |
| Base Media | DMEM-LG, Euro-Collins solution, University of Wisconsin solution [41] [30] | Provide physiological ion balance, nutrient support during vitrification process |
| Specialized Devices | Nylon membrane carriers [41], MTG directional freezers [38], droplet generators [39] | Enable ultra-rapid cooling rates, precise thermal control |
| Assessment Tools | Membrane integrity stains, flow cytometry panels, differentiation assays [41] [30] | Post-thaw viability and functionality assessment |
| Cold Chain Equipment | Liquid nitrogen storage systems, controlled-rate freezers, portable cryoshippers | Maintain stable cryogenic temperatures for long-term storage |
Advanced Applications in MSC-Based Tissue Engineering
The application of vitrification techniques extends beyond simple MSC suspensions to complex tissue-engineered constructs requiring preservation of spatial organization and cell-matrix interactions. For three-dimensional MSC constructs such as spheroids, hydrogels, or scaffold-based systems, equilibrium vitrification typically yields superior outcomes by maintaining structural integrity [30]. The controlled CPA exposure minimizes matrix disruption while still providing sufficient dehydration for successful vitrification.
Recent innovations in vitrification technology show particular promise for MSC-based therapeutics. Directional freezing techniques, which move samples through precisely controlled thermal gradients, enable improved vitrification of larger constructs by managing ice crystal propagation directionality [38]. Droplet-based vitrification approaches, inspired by bioprinting technologies, encapsulate MSC aggregates in minimal fluid volumes before ultra-rapid cooling, achieving exceptional cooling rates while minimizing CPA requirements [39].
Figure 2: Molecular mechanisms of cryoprotectant action during vitrification, illustrating the pathway from CPA introduction to glass transition achievement.
For clinical translation of MSC-based therapies, vitrification protocols must address regulatory requirements including standardized procedures, defined composition CPAs, and comprehensive quality assessment. The development of serum-free, xeno-free CPA formulations represents a critical advancement toward clinical-grade vitrification protocols [30]. Additionally, implementing rigorous post-thaw assessment protocols that evaluate not only viability but also differentiation potential, immunomodulatory capacity, and secretome profile ensures the functional preservation of vitrified MSC products [4] [30].
Vitrification techniques offer powerful approaches for preserving MSC-based tissue-engineered structures, with equilibrium and non-equilibrium methods providing complementary advantages for different applications. Equilibrium vitrification excels with larger, complex constructs where controlled osmotic processes maintain structural integrity, while non-equilibrium approaches provide superior ice inhibition for smaller volumes where ultra-rapid cooling is achievable. The continued refinement of vitrification protocols, coupled with advances in CPA formulations and thermal management technologies, will further enhance the preservation efficacy for these clinically relevant cellular therapeutics. As the field progresses, standardized vitrification workflows will play an increasingly vital role in enabling off-the-shelf availability of MSC-based products for regenerative medicine and drug development applications.
Cryoprotectant Agents (CPAs) are fundamental to the field of cryobiology, enabling the preservation of biological materials at ultralow temperatures by mitigating the damaging effects of ice crystal formation and osmotic stress [34]. For research focused on mesenchymal stem cell (MSC)-based tissue-engineered structures, the selection of an appropriate CPA is not merely a procedural step but a critical determinant of post-thaw cell viability, functionality, and the structural integrity of complex constructs [30]. CPAs are broadly categorized into two classes based on their ability to cross cell membranes: permeating and non-permeating agents.
Permeating CPAs, such as dimethyl sulfoxide (DMSO) and glycerol, are characterized by their low molecular weight and ability to diffuse across the plasma membrane. Their primary mechanism of action involves replacing water molecules within the cell, thereby depressing the freezing point and reducing the amount of intracellular ice that forms during cooling [34] [30]. Non-Permeating CPAs, including sugars like sucrose and trehalose, are typically larger molecules that do not enter the cell. They function extracellularly by inducing osmotic dehydration of the cell prior to freezing, reducing the potential for lethal intracellular ice formation, and promoting vitrification—the transition of water into a glassy, non-crystalline state [34] [43]. The strategic use of these agents, either alone or in combination, forms the cornerstone of effective cryopreservation protocols for advanced regenerative medicine applications.
Core Mechanisms of Action
Physical and Chemical Principles
The injury to cells during freezing primarily stems from two phenomena: the mechanical damage caused by intracellular and extracellular ice crystals, and the deleterious increase in solute concentration in the remaining liquid phase as pure water freezes out [34]. CPAs counteract these processes through several interconnected mechanisms.
Permeating CPAs like DMSO and glycerol readily penetrate the cell. Their relatively small size (typically less than 100 daltons) and amphiphilic nature allow them to cross the lipid bilayer [34]. Once inside, they form strong hydrogen bonds with water molecules, effectively depressing the freezing point of the intracellular solution and reducing the quantity of water available to form ice nuclei. At a commonly used concentration of 10%, DMSO is also known to increase membrane porosity, facilitating water movement out of the cell and further preventing intracellular ice formation [34]. However, at high concentrations, these agents can become toxic; for instance, DMSO at 40% concentration can cause lipid bilayers to disintegrate [34].
Non-permeating CPAs, such as trehalose and sucrose, exert their protective effect from outside the cell. They are kosmotropes, meaning they order the surrounding water molecules, which alters the hydrogen bond network and inhibits ice crystal growth [43]. By increasing the osmolarity of the extracellular solution, they draw water out of the cell, leading to protective dehydration. Furthermore, according to the "water replacement hypothesis," these sugars can stabilize phospholipids and proteins by hydrogen bonding to them, effectively replacing the water molecules that are normally bound in a hydrated state, thus preserving structural integrity during dehydration and freezing [43].
Table 1: Fundamental Mechanisms of Permeating vs. Non-Permeating Cryoprotectants
| Feature | Permeating CPAs (DMSO, Glycerol) | Non-Permeating CPAs (Trehalose, Sucrose) |
|---|---|---|
| Cellular Interaction | Cross the cell membrane [30] | Remain outside the cell [30] |
| Primary Intracellular Action | Depress freezing point, reduce intracellular ice formation [34] | Induce protective cell dehydration via osmosis [34] |
| Primary Extracellular Action | Reduce water activity in extracellular space [44] | Promote vitrification, inhibit ice recrystallization [43] [44] |
| Membrane Interactions | Can increase membrane permeability at specific concentrations [34] | Stabilize membrane via water replacement hypothesis [43] |
| Typical Concentrations | DMSO: ~10% (2M); Glycerol: 10-30% [34] [45] | Trehalose: 100-400 mM [43] |
Application Notes for MSC and Tissue-Engineered Structures
The cryopreservation of MSC-based tissue-engineered structures presents unique challenges beyond those of cell suspensions. The three-dimensional architecture can impose physical barriers to the uniform penetration of CPAs and create complexities in heat transfer during cooling and warming [30]. Success hinges on optimizing CPA cocktails and protocols to protect not only individual cells but also the extracellular matrix and cell-matrix interactions.
CPA Cocktail Formulations and Efficacy
Combining permeating and non-permeating CPAs often yields synergistic benefits. The non-permeating agent can reduce the required concentration of the more toxic permeating CPA, while the permeating agent ensures intracellular protection that the sugar alone cannot provide [34] [45].
Table 2: Exemplary CPA Formulations for MSC Cryopreservation
| Cell Type / Structure | CPA Formulation | Cooling Rate | Reported Outcome | Citation |
|---|---|---|---|---|
| Adipose-Derived Stem Cells (ADSCs) | 1.0 M Trehalose + 20% Glycerol | 1°C/min | Similar viability to 10% DMSO + 90% FBS, higher migration capability [45] | [45] |
| Human Pluripotent Stem Cells | 500 mM Trehalose + 10% Glycerol | Not Specified | 20-30% increase in relative viability vs. 10% DMSO; phenotype maintained [43] | [43] |
| Mesenchymal Stem Cells (General) | 10% DMSO + non-permeating agent (e.g., sucrose) | ~1°C/min | Common baseline protocol; slow cooling recommended for hepatocytes, hematopoietic & mesenchymal stem cells [34] | [34] |
| Human Umbilical Cord Blood Stem Cells | 146 mM Trehalose + 5-10% DMSO | Not Specified | Proliferation as high as non-frozen control group [43] | [43] |
| Murine Spermatogonial Stem Cells | 50 mM Trehalose + 10% DMSO | Not Specified | Improved viability (90% vs. 76%) after one week of storage [43] | [43] |
The Scientist's Toolkit: Essential Reagents for Cryopreservation
Table 3: Key Research Reagent Solutions for Cryopreservation Protocols
| Reagent / Material | Function / Application | Notes |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating CPA; standard for many cell types including MSCs [34] [30]. | Use high purity grade. Can be toxic at high concentrations; stepwise addition is recommended near 0°C to minimize toxicity [34]. |
| Glycerol | Permeating CPA; often used as a less toxic alternative to DMSO [30] [45]. | Common in combinations with trehalose for xeno-free formulations [45]. |
| Trehalose | Non-permeating disaccharide; stabilizes membranes and promotes vitrification [43] [45]. | Optimal concentration is often 100-400 mM; higher concentrations can cause osmotic damage [43]. |
| Sucrose | Non-permeating disaccharide; used as an extracellular CPA and osmotic buffer [4]. | Frequently used in vitrification mixtures and for diluting CPAs during thawing to prevent osmotic shock. |
| Polyvinylpyrrolidone (PVP) | High molecular weight, non-permeating polymer CPA [34] [44]. | Functions as an ice recrystallization inhibitor; does not enter the cell. |
| Cryo-Media Base (e.g., FBS, Xeno-Free Alternatives) | Provides a supportive, nutrient-rich base for CPA solutions. | FBS is common but poses zoonotic risk; clinical applications are moving toward xeno-free defined media [45]. |
| Programmable Freezer / Mr. Frosty | Controls cooling rate for slow freezing protocols (~1°C/min) [45] [4]. | Critical for reproducible slow freezing. Mr. Frosty uses isopropanol to approximate a -1°C/min cooling rate. |
| Liquid Nitrogen Storage System | Long-term storage of cryopreserved samples at -196°C [4]. | Samples can be stored in liquid or vapor phase; vapor phase reduces risk of liquid nitrogen ingress into vials. |
Detailed Experimental Protocols
Protocol: Cryopreservation of MSCs with a Trehalose-Glycerol Combination
This protocol is adapted from a study demonstrating successful cryopreservation of human Adipose-Derived Stem Cells (ADSCs) using a combination of 1.0 M trehalose and 20% glycerol, resulting in post-thaw viability and function comparable to, and in some aspects superior to, standard DMSO-based methods [45].
I. Pre-Cryopreservation
- CPA Preparation: Prepare the cryoprotective solution: 1.0 M trehalose and 20% (v/v) glycerol in phosphate-buffered saline (PBS). Filter-sterilize using a 0.22-μm filter.
- Cell Preparation: Culture ADSCs (or other MSCs) to 80-90% confluency at passage 3. Wash the cells with PBS and detach using 0.25% trypsin-EDTA. Neutralize the trypsin with culture medium containing serum.
- Cell Counting and Centrifugation: Count the cells and centrifuge the suspension at 1500 rpm for 5 minutes. Resuspend the cell pellet in the prepared 1.0 M Tre + 20% Gly CPA solution to a final concentration of 1 × 10^6 cells per milliliter.
II. Freezing Process
- Aliquoting: Transfer 1 ml of the cell suspension into labeled cryovials.
- Slow Freezing: Place the cryovials immediately into a Nalgene Mr. Frosty freezing container or a programmable freezer, pre-cooled to 4°C.
- Controlled Cooling: Freeze the cells at a controlled cooling rate of -1°C per minute to -80°C. Store the vials at -80°C overnight.
- Long-Term Storage: The following day, transfer the cryovials to a liquid nitrogen storage tank (-196°C) for long-term preservation.
III. Post-Thaw & Analysis
- Thawing: Rapidly thaw the cryovials by placing them in a 37°C water bath with gentle agitation until the ice is completely melted.
- CPA Removal: To remove the CPA, dilute the thawed cell suspension in 10 ml of pre-warmed PBS or culture medium. Centrifuge at 1500 rpm for 5 minutes. Repeat this washing step once more.
- Viability Assessment: Resuspend the final cell pellet in culture medium. Assess cell viability using Trypan Blue exclusion assay and an automated cell counter.
- Functional Assays (Recommended): Plate the cells for subsequent functional assays to ensure cryopreservation has not compromised key properties:
- Proliferation: Use a CCK-8 assay at 24, 48, and 72 hours.
- Migration: Perform a scratch/wound healing assay over 24 hours.
- Stemness and Differentiation: Analyze surface markers via flow cytometry and conduct tri-lineage (osteogenic, adipogenic, chondrogenic) differentiation assays.
Protocol: Proteomic Evaluation of CPA Efficacy
This protocol outlines a proteomics-based approach to evaluate the molecular impact of different CPA formulations on yeast (S. cerevisiae), a method that can be adapted for MSC-based research to gain deep mechanistic insights [44].
I. Treatment and Freezing
- Inoculum Prep: Grow S. cerevisiae to mid-log phase (OD600 ~0.80) in Yeast Malt (YM) broth.
- CPA Application: Combine the inoculum with an equal volume of the designated CPA treatment (e.g., DMSO alone, glycerol alone, trehalose alone, or various combinations) in cryovials.
- Controlled-Rate Freezing: Freeze the samples using a controlled-rate freezer. Cool from room temperature to -40°C at -1°C/min, then ramp down to -90°C at -10°C/min. Transfer vials to -80°C for storage (e.g., 1 week).
II. Post-Thaw Analysis
- Viability Testing: Thaw cryovials in a 37°C water bath. Perform serial dilutions and spot 4 µL of each dilution onto YM agar plates. Incubate at 28°C for 24 hours and count colonies to determine recovery rates.
- Protein Extraction: Thaw parallel vials and centrifuge to pellet cells. Wash pellets with chilled sterile water. Lyse cells using a commercial lysis reagent (e.g., CelLytic Y) supplemented with 5 mM dithiothreitol (DTT). Clarify the lysate by centrifugation and store the supernatant (crude protein extract) at -80°C.
III. LC-MS/MS and Data Analysis
- Sample Preparation: Measure protein concentration using a BCA assay. Digest 100 µg of protein from each sample with trypsin.
- TMT Labeling: Label the resulting peptides from different CPA treatment groups using Tandem Mass Tag (TMT) reagents (e.g., TMT-18plex) as per manufacturer's instructions.
- LC-MS/MS: Analyze the pooled, labeled peptides by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS).
- Bioinformatics: Identify and quantify proteins using database search software. Perform functional analysis (e.g., KEGG pathway enrichment) on proteins that are significantly upregulated or downregulated in each CPA formulation group compared to controls to understand the specific stress responses and protective mechanisms invoked.
Visualization of Protocols and Pathways
Experimental Workflow for CPA Evaluation
The following diagram illustrates the integrated workflow for evaluating cryoprotectant agents, from preparation to functional and omics-based analysis.
CPA Mechanisms and Decision Logic
This flowchart outlines the logical decision-making process for selecting and combining cryoprotectant agents based on their mechanisms and the requirements of the biological sample.
The therapeutic potential of Mesenchymal Stem Cells (MSCs) in regenerative medicine is undeniable, with applications spanning from orthopedic injuries to autoimmune diseases [1]. A critical component for the clinical translation of MSC-based therapies is effective cryopreservation, which enables long-term storage, quality control, and the creation of "off-the-shelf" cell products [4] [46]. However, the post-thaw recovery process, encompassing warming and cryoprotective agent (CPA) removal, presents a significant bottleneck. Inefficient or damaging protocols can severely compromise cell viability, functionality, and the overall therapeutic efficacy of the final product [46] [30].
This Application Note provides detailed, standardized protocols for the thawing and post-thaw processing of cryopreserved MSCs and MSC-based tissue-engineered structures. The procedures outlined herein are designed to minimize osmotic injury and preserve critical cellular functions, such as immunomodulation and differentiation potential, which are essential for successful clinical outcomes [1] [47]. By establishing a robust and reproducible workflow from vial removal to ready-to-use cells, this document aims to support researchers and therapy developers in enhancing the safety, efficacy, and standardization of advanced MSC-based therapeutic products.
Background and Significance
Cryopreservation is the cornerstone for biobanking MSCs, allowing for the preservation of living cells at ultra-low temperatures (typically -196°C in liquid nitrogen) [4] [30]. The process relies on CPAs to protect cells from the lethal damage associated with intracellular ice formation and excessive solute concentration. The two primary cryopreservation methods are slow freezing and vitrification, each with distinct mechanisms and implications for the thawing process [4].
Slow freezing, the most common method for MSC suspension storage, involves a controlled, slow cooling rate that promotes cellular dehydration, minimizing intracellular ice [4]. Vitrification, an alternative approach, uses high CPA concentrations and ultra-rapid cooling to solidify cells and their environment into a glassy, non-crystalline state [4]. While the cooling methods differ, the thawing phase is critical for both. The primary goals of any thawing protocol are to:
- Rapidly melt the frozen sample to avoid the damaging effects of ice recrystallization during warming [48].
- Safely and efficiently remove CPAs to mitigate their chemical toxicity and osmotic shock [4] [49].
- Maximize the recovery of viable, functional cells capable of executing their intended therapeutic mechanism of action [46].
The transition of MSCs from research tools to clinically approved drugs, such as Cartistem for osteoarthritis, underscores the urgent need for standardized, GMP-compliant post-thaw protocols to ensure product consistency and patient safety [47].
Thawing and CPA Removal: Core Principles and Workflow
The following section details the core procedural steps, from retrieving the cryovial to preparing the cells for their final application. A comprehensive overview of this workflow is presented in Figure 1.
Rapid Warming Protocol
The established standard for thawing cryopreserved MSC suspensions is rapid warming in a 37°C water bath. This method is designed to quickly traverse the dangerous temperature zone where ice recrystallization can cause significant mechanical damage to cells [48].
Materials:
- Pre-warmed water bath (37°C)
- Personal protective equipment (PPE): lab coat, gloves, face shield
- Sterile tissue or wipes
- 70% ethanol spray or wipe
- Cryovial(s) containing cryopreserved MSCs
Procedure:
- Safety Precautions: Don appropriate PPE. Ensure all procedures are performed in a certified biological safety cabinet to maintain sterility.
- Bath Preparation: Verify that the water bath is clean and maintained at 37°C. For GMP-comformant processes, use a dry-warming device or a closed, sterile water bath system to eliminate the risk of microbial contamination from the water [4] [48].
- Vial Retrieval and Transfer: Quickly retrieve the cryovial from the liquid nitrogen storage tank and immediately transport it to the water bath. Minimize the time the vial spends above -130°C to prevent detrimental physicochemical changes.
- Agitation for Efficient Heat Transfer: Submerge the vial in the 37°C water bath and gently agitate it continuously until only a small ice crystal sliver remains (typically 2-3 minutes). The warming rate should exceed 100°C/min where possible [4].
- Decontamination: Carefully wipe the outside of the vial with a sterile wipe soaked in 70% ethanol before introducing it into the biological safety cabinet.
Table 1: Impact of Cooling and Thawing Rates on T Cell Viability (as a model somatic cell)
| Cooling Rate (°C/min) | Thawing Rate (°C/min) | Relative Viable Cell Number | Key Observation |
|---|---|---|---|
| -1 (Slow) | 113 (Rapid) | High | No significant impact of warming rate observed. |
| -1 (Slow) | 1.6 (Very Slow) | High | Viability maintained with slow cooling. |
| -10 (Rapid) | 113 (Rapid) | High | Rapid warming rescues viability after rapid cooling. |
| -10 (Rapid) | 6.2 (Slow) | Significantly Reduced | Ice recrystallization causes mechanical cell damage. |
Note: Data adapted from a study on human peripheral blood T cells, demonstrating the critical interaction between cooling and warming rates [48]. For slowly frozen MSCs, rapid warming is consistently recommended.
CPA Removal via Dilution-Centrifugation
Following thawing, the removal of CPAs, particularly Dimethyl Sulfoxide (DMSO), is essential due to its potential toxicity to both cells and patients [4] [30]. The conventional method is dilution followed by centrifugation.
Materials:
- Complete cell culture medium (e.g., basal medium with serum or serum-free supplements)
- Sterile centrifuge tubes
- Centrifuge
- Pipettes and sterile tips
Procedure:
- Controlled Dilution: Gently transfer the thawed cell suspension from the cryovial into a larger volume (e.g., 10-fold) of pre-warmed, complete culture medium. This gradual dilution reduces the extracellular CPA concentration slowly, minimizing the osmotic pressure difference that drives excessive water into the cells and causes swelling and potential lysis [4] [49].
- Centrifugation: Centrifuge the cell suspension at a moderate speed (e.g., 300-400 x g) for 5-10 minutes to pellet the cells.
- Supernatant Aspiration: Carefully aspirate and discard the supernatant, which contains the diluted CPAs.
- Cell Resuspension: Gently resuspend the cell pellet in fresh, pre-warmed complete culture medium to the desired concentration for counting, viability assessment, or direct application.
Table 2: Common Cryoprotective Agents (CPAs) and Their Properties
| CPA | Type | Common Concentration | Key Considerations |
|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating | 5-10% | Gold standard; can be cytotoxic and trigger allergic reactions [4]. |
| Glycerol | Penetrating | 5-10% | Lower toxicity but less effective for some cell types [4] [30]. |
| Ethylene Glycol (EG) | Penetrating | - | Lower cell toxicity than DMSO but similar cryopreservation effect [4]. |
| Sucrose | Non-penetrating | 0.1-0.5 M | Used as an osmotic buffer; often combined with penetrating CPAs [30]. |
| Trehalose | Non-penetrating | 0.1-0.5 M | Stabilizes cell membranes; often used in combination [30]. |
Figure 1. Standardized workflow for thawing and CPA removal of cryopreserved MSC suspensions. The process emphasizes rapid warming and controlled dilution to maximize cell recovery. BSC: Biological Safety Cabinet.
Advanced and Alternative CPA Removal Methodologies
While dilution-centrifugation is widely used, it has drawbacks, including cell clumping, loss during handling, and shear stress [49]. For sensitive cell types or larger volumes, advanced methods are emerging.
Dilution-Filtration Systems
These systems use a hemodialyzer or hemofilter in a closed-loop circuit to continuously dilute and remove CPAs while concentrating the cells, thereby reducing manual handling and osmotic stress [50] [49].
Key Advantage: The extracellular CPA concentration can be decreased more gradually and continuously than in a single-step dilution, allowing cells more time to equilibrate and reducing volume excursions [50].
Experimental Protocol (Based on a 3-in-1 Multifunctional System):
- Setup: The thawed cell suspension is placed in a closed-loop system with a peristaltic pump and a hemodialyzer (e.g., Plasmflo AP-05H/L) [50] [49].
- Circulation and Dilution: The cell suspension is circulated while a diluent (e.g., isotonic saline or a hypertonic solution to control swelling) is introduced at a controlled flow rate.
- Filtration: The solution containing CPAs is filtered out through the hollow fibers of the dialyzer, while the cells are retained in the circulating loop.
- Concentration: The process continues until the target CPA concentration is achieved, resulting in a washed and concentrated cell product ready for use [49].
Optimization: Computer-guided protocols that incorporate cell-type-specific parameters (cell membrane permeability to water and CPA) can optimize the diluent flow rate to minimize processing time and osmotic injury [50] [49].
The Scientist's Toolkit: Essential Reagents and Equipment
Table 3: Key Research Reagent Solutions and Equipment
| Item | Function/Description | Example/Catalog Consideration |
|---|---|---|
| Cryopreservation Medium | Formulation containing CPAs to protect cells during freezing. | e.g., Commercial CryoStor CS10 [48] or lab-made (e.g., 90% FBS + 10% DMSO). |
| Complete Culture Medium | Base medium for diluting thawed cells and post-thaw culture. | e.g., α-MEM or DMEM, supplemented with FBS and growth factors. |
| DMSO (GMP-grade) | Penetrating CPA; the most commonly used for MSCs. | Sourced from GMP-compliant manufacturers to ensure quality and traceability. |
| Sucrose/Trehalose | Non-penetrating CPAs; used as osmotic buffers in combination. | High-purity, sterile-filtered solutions. |
| Water Bath or Dry Warmer | Device for achieving rapid, controlled warming at 37°C. | Dry warming systems are preferred for cGMP compliance to avoid contamination [4] [48]. |
| Hemofilter / Hemodialyzer | Core component of dilution-filtration systems for CPA removal. | e.g., Asahi Kasei Plasmflo or Fresenius Hemofilter models [50] [49]. |
| Peristaltic Pumps | For driving fluid flow in automated dilution-filtration systems. | Pumps capable of precise flow rate control for reproducible protocols. |
Post-Thaw Assessment and Analysis
A comprehensive assessment of cell quality after thawing and CPA removal is non-negotiable. Key parameters to evaluate include:
- Viability: Use trypan blue exclusion or more sensitive flow cytometry-based assays (e.g., LIVE/DEAD staining with propidium iodide) [48]. A viability of >70-80% is typically expected for slow-frozen MSCs [4].
- Cell Count and Recovery: Determine the total and viable cell number to calculate recovery efficiency.
- Phenotype: Verify the expression of characteristic MSC surface markers (CD73, CD90, CD105) and the absence of hematopoietic markers (CD34, CD45) via flow cytometry to ensure identity is retained post-thaw [1] [47].
- Functionality:
- Proliferation Potential: Monitor growth kinetics over several days.
- Clonogenic Capacity: Assess using Colony-Forming Unit Fibroblast (CFU-F) assays.
- Differentiation Potential: Confirm multilineage capacity by inducing osteogenic, adipogenic, and chondrogenic differentiation [1].
- Immunomodulatory Activity: Measure the secretion of key bioactive molecules (e.g., PGE2, TSG-6, IDO) or the ability to suppress the activation of immune cells like T-cells in co-culture assays [1] [47].
The transition of MSC therapies from the laboratory to the clinic hinges on robust and reproducible manufacturing processes, with cryopreservation and thawing being critical control points. The standardized protocols detailed in this document—emphasizing rapid warming, controlled CPA removal, and rigorous post-thaw assessment—provide a foundational framework for researchers and therapy developers. By adopting these best practices and staying abreast of technological advancements like automated dilution-filtration systems, the field can enhance the consistency, quality, and ultimate success of MSC-based regenerative medicines.
Within the broader context of research on mesenchymal stem cell (MSC)-based tissue-engineered structures, effective post-thaw processing is not merely a final step but a critical determinant of experimental reproducibility and therapeutic efficacy. Cryopreservation induces significant stress on cells, and for complex tissue-engineered constructs, the challenges are magnified due to the need to preserve both cell viability and the integrity of the three-dimensional microstructure [30]. The immediate post-thaw phase is a vulnerable period where cells are subjected to osmotic stress, cryoprotectant agent (CPA) toxicity, and the potential damage from ice crystal formation [51] [30]. Therefore, the protocols for centrifugation, cell washing, and viability assessment are designed to mitigate these stresses, efficiently remove toxic CPAs like DMSO, and accurately evaluate the success of the cryopreservation process. This document provides detailed application notes and protocols for these critical post-thaw procedures, specifically framed for researchers working with MSC-based tissue-engineered products.
The Critical Post-Thaw Workflow
The journey from a cryopreserved vial to a viable, functional cell population ready for experimentation or implantation involves a series of interconnected steps. The following workflow diagrams the core post-thaw process and the key decision points for optimizing viability and recovery, particularly for sensitive MSC-based constructs.
Detailed Experimental Protocols
Thawing and Initial Dilution
The initial thawing and dilution steps are time-sensitive and critical for minimizing the cytotoxic effects of cryoprotectants like DMSO.
Materials:
- Pre-warmed complete culture medium (37°C)
- Water bath (37°C, calibrated)
- 70% ethanol spray
- Centrifuge tubes (e.g., 15 mL or 50 mL conical tubes)
Method:
- Rapid Thawing: Remove the cryovial from liquid nitrogen storage and immediately place it in a 37°C water bath. Gently agitate the vial until only a small ice crystal remains (approximately 1-2 minutes). Do not completely thaw the vial at room temperature [51] [52] [53].
- Decontamination: Wipe the exterior of the cryovial thoroughly with 70% ethanol and transfer it to a biological safety cabinet [53].
- Immediate Dilution: Gently transfer the entire contents of the cryovial into a centrifuge tube containing a pre-warmed dilution medium. A 1:10 dilution is recommended; for example, add 1 mL of cell suspension to 9 mL of medium. This step rapidly reduces the concentration of DMSO, mitigating its toxicity and osmotic shock [52] [54] [53]. Gently pipette to mix.
Centrifugation and Cell Washing
This step aims to pellet the cells and remove the cryopreservation medium containing the cryoprotectant.
Materials:
- Bench-top centrifuge
- Pre-warmed complete culture medium or PBS (without Ca2+/Mg2+)
Method:
- Centrifugation: Centrifuge the diluted cell suspension at 300 x g for 5 minutes at room temperature (20-25°C). Avoid using a refrigerated centrifuge set to 4°C, as cold temperatures can exacerbate cell stress [52] [54].
- Supernatant Removal: Carefully decant the supernatant without disturbing the cell pellet. The supernatant contains the spent cryopreservation medium and most of the DMSO.
- Cell Washing: Gently resuspend the cell pellet in a fresh volume of pre-warmed culture medium or PBS. Repeat the centrifugation step (300 x g for 5 minutes) and carefully remove the supernatant. A single wash is typically sufficient, but for therapeutic applications or if residual CPA is a concern, a second wash may be performed [51] [55].
Viability and Cell Count Assessment
Accurate assessment of post-thaw viability is essential for standardizing subsequent experiments, such as seeding densities for 3D constructs.
Materials:
- Hemocytometer or automated cell counter
- Trypan blue solution (0.4%) or other viability dyes (e.g., acridine orange/propidium iodide)
- PBS
Method:
- Resuspension: After the final wash, resuspend the cell pellet in an appropriate, known volume of PBS or culture medium.
- Staining: Mix a small aliquot of the cell suspension with an equal volume of trypan blue solution. Incubate for 1-2 minutes. Viable cells will exclude the dye, while non-viable cells with compromised membranes will take up the blue stain [52] [54].
- Counting: Load the mixture onto a hemocytometer and count the cells under a microscope. Alternatively, use an automated cell counter for improved speed and reproducibility.
- Calculation:
- Total Cell Count: Calculate the concentration of all cells (stained and unstained) per mL.
- Viability Percentage: (Number of unstained cells / Total number of cells) x 100.
Table 1: Standard Parameters for Post-Thaw Processing Steps
| Processing Step | Key Parameters | Rationale & Considerations | Primary Reference |
|---|---|---|---|
| Thawing & Dilution | 37°C water bath; 1:10 dilution with pre-warmed media | Rapid thawing prevents ice recrystallization damage. Immediate dilution reduces CPA toxicity and osmotic stress. | [51] [52] [53] |
| Centrifugation | 300 x g for 5 minutes at 20-25°C | Gentle spin pellets cells without causing excessive mechanical stress. Room temperature avoids cold-induced shock. | [52] [54] |
| Washing | One to two washes with culture media/PBS | Ensures effective removal of cytotoxic CPAs like DMSO. Over-washing may lead to mechanical cell loss. | [51] [55] |
| Viability Assessment | Trypan Blue exclusion; Hemocytometer/Automated Counter | Provides a quick and standardized measure of post-thaw cell health. Correlates with initial recovery potential. | [52] [54] |
Quantitative Data and Method Comparison
The choice of post-thaw processing method involves trade-offs between cell recovery, purity, and functional fitness. These trade-offs are particularly important when processing complex starting materials like cord blood units, which share similarities with tissue-engineered constructs in terms of cellular heterogeneity.
Table 2: Impact of Post-Thaw Processing Methods on Cell Recovery and Function
| Processing Method | Impact on Cell Recovery (Yield) | Impact on Purity | Impact on Functional Fitness | Best Suited For |
|---|---|---|---|---|
| Wash-Only | Highest yield retained. | Lowest purity levels; retains contaminants like platelets and debris. | Viability can be maintained but function may be affected by contaminants. | Applications where maximizing absolute cell number is the priority over purity. |
| Density Gradient | Intermediate recovery; some cell loss during separation. | Effective removal of red blood cells and dead cells/granulocytes. | Good preservation of function; clean population for downstream work. | Standard purification of mononuclear cells from heterogeneous samples. |
| Immunodepletion (Beads) | Lower recovery due to specific cell removal. | Highest purity for target population. | Best preserved viability over 5 days of culture; ideal for long-term assays. | Applications requiring a highly specific, pure cell population. |
| PBMC Isolation Kit | Good recovery of target cells. | High depletion efficiency; may significantly alter subset composition (e.g., CD14+). | High viable cell percentage on Day 0; may reduce specific functions (e.g., T cell proliferation). | Rapid isolation of a defined cell population with consistent initial viability. |
Data adapted from a study on cord blood mononuclear cell processing, demonstrating the application-specific trade-offs in method selection [56] [57].
The Scientist's Toolkit: Essential Reagents and Materials
Successful post-thaw processing relies on a suite of reliable reagents and equipment. The following toolkit details essential items for these critical protocols.
Table 3: Research Reagent Solutions for Post-Thaw Processing
| Item | Function/Application | Specific Examples & Notes |
|---|---|---|
| Cryopreservation Media | Provides a protective environment during freezing/thawing. Contains CPAs. | CryoStor CS10: A ready-to-use, serum-free formulation [55]. Lab-made media: Often 90% FBS + 10% DMSO, but has variability concerns [55] [52]. |
| Basal Dilution/Wash Media | Dilutes cryopreservation medium and washes cells post-centrifugation. | RPMI 1640, DMEM, IMDM supplemented with 10-20% FBS. Serum helps stabilize cells and mitigate osmotic stress [55] [53]. |
| Cryoprotectant Agents (CPAs) | Protect cells from ice crystal damage during freezing. | DMSO (Intracellular): Penetrates cell membrane; can be cytotoxic [51] [30]. Sucrose/Trehalose (Extracellular): Do not penetrate; help stabilize cell membranes [51] [30]. Polyampholytes (Synthetic): Emerging macromolecular CPAs that reduce intracellular ice formation [54]. |
| Viability Stain | Differentiates live from dead cells for counting. | Trypan Blue (0.4%): Standard dye exclusion method [52] [54]. Acridine Orange/Propidium Iodide (AO/PI): Used in automated cell counters for higher accuracy. |
| Specialized Centrifuge Tubes | Safe containment of cells during centrifugation steps. | 15 mL and 50 mL conical tubes. Use sterile, validated tubes to prevent leakage or contamination. |
Optimizing for MSC-Based Tissue-Engineered Structures
Applying these protocols to MSC-based tissue-engineered structures (e.g., cells seeded on 3D scaffolds) introduces additional complexity. The primary challenge is that the structure itself must often be handled as a single unit, making traditional centrifugation and washing difficult.
Key Considerations:
- Handling 3D Constructs: Washing may involve gentle gravity flow or perfusion of the scaffold with warm culture medium to slowly displace the cryoprotectant, rather than centrifugation [30].
- Viability Assessment: Standard trypan blue counting requires dissociating cells from the scaffold, which itself can cause significant cell death and yield inaccurate results. Alternative methods like Live/Dead staining (calcein AM/ethidium homodimer) followed by confocal microscopy and image analysis are required to assess viability in situ within the 3D structure [30].
- Functional Assessment: For tissue-engineered products, viability alone is an insufficient metric. Post-thaw functionality must be confirmed through assays of MSC-specific properties, such as trilineage differentiation potential, immunomodulatory capacity, and secretory profile [25] [30].
The following diagram outlines the critical decision pathway for selecting the appropriate viability assessment method based on the nature of the cryopreserved product.
The clinical translation of mesenchymal stem cell (MSC)-based tissue-engineered structures hinges on the development of robust preservation technologies that ensure product viability, functionality, and immediate availability. Cryopreservation serves as a pivotal biomanufacturing step enabling long-term storage and forming the foundation of "off-the-shelf" tissue products for regenerative medicine [58] [30]. This application note details standardized protocols and workflows for the cryopreservation of two distinct tissue-engineered structures: scaffold-based constructs and scaffold-free pre-differentiated tissues. These protocols are designed to address the critical challenge of maintaining cellular viability, differentiation potential, and structural integrity post-thaw, thereby supporting reproducible research and clinical applications.
Cryopreservation of Scaffold-Based Cell Constructs
Three-dimensional scaffolds provide a structural template that guides cell growth, organization, and tissue formation. Their porous architecture presents unique challenges and opportunities during cryopreservation.
Experimental Protocol: Cryopreservation of Cell-Seeded Porous Scaffolds
Materials:
- Porous Scaffolds: SPCL (starch and polycaprolactone blend) fiber meshes (8 mm diameter × 3.5-4 mm thickness) [58]
- Cells: Goat bone marrow stromal cells (GBMSCs) at passage 4 [58]
- Cryoprotective Solution: 10% Dimethyl Sulfoxide (DMSO) in Fetal Bovine Serum (FBS) [58]
- Equipment: Cryovials, controlled-rate freezer, liquid nitrogen tank, 24-well nonadherent plates, water bath [58]
Methodology:
- Scaffold Preparation and Seeding: Sterilize SPCL scaffolds using ethylene oxide. Seed scaffolds with GBMSCs at a density of 5×10^5 cells in 300 µL volume. Transfer carefully to 24-well nonadherent plates [58].
- Pre-cryopreservation Culture: Incubate seeded constructs for 3 hours before adding 1.5 mL of low-glucose DMEM supplemented with 10% FBS and 1% antibiotic/antimycotic. Culture for 7 days, changing medium every 2-3 days [58].
- Cryopreservation: After 7 days of culture, suspend constructs in cryoprotective solution (10% DMSO in FBS) inside standard cryovials. Use a controlled-rate freezing system and store in liquid nitrogen (-196°C) for at least 7 days [58].
- Thawing and Recovery: Partially thaw constructs in a 37°C water bath. Transfer to 24-well nonadherent plates and add cold DMEM basal medium with 20% FBS. Culture for 9 days post-thaw, changing medium every 2-3 days to allow cellular recovery and DMSO leaching [58].
Table 1: Viability Assessment of Cryopreserved Scaffold-Based Constructs
| Assessment Method | Pre-cryopreservation Results | Post-thaw/Recovery Results | Key Findings |
|---|---|---|---|
| MTS Assay | Baseline viability established | High viability retention | Normalized to scaffold surface area [58] |
| DNA Quantification | Cellular content baseline | Maintained DNA content | Indicates cell retention within scaffold [58] |
| SEM Analysis | Confluent cell layer on scaffold | Preserved cell-scaffold integration | Demonstrates structural integrity [58] |
| Micro-CT | Scaffold architecture baseline | Maintained porosity & interconnectivity | Confirms architectural preservation [58] |
DMSO-Free Cryopreservation Approach for 3D Bioprinted Constructs
Recent advances in bioink development enable cryopreservation with reduced cytotoxic cryoprotectants:
Materials:
- Bioink: Alginate-nanocellulose hybrid incorporating Hyaluronic Acid (Ha) [59]
- Cryopreservation: Ultra-low temperature storage without DMSO [59]
Methodology:
- Bioink Preparation: Formulate alginate-nanocellulose bioink with incorporated hyaluronic acid [59].
- 3D Bioprinting: Fabricate scaffolds using the enhanced bioink [59].
- Cryopreservation: Cryopreserve cell-laden scaffolds at ultra-low temperatures without DMSO addition [59].
- Post-thaw Analysis: Assess viability, metabolic activity, and scaffold properties (porosity, surface roughness, mechanical durability) [59].
Key Findings: Hyaluronic acid incorporation improves bioink viscoelasticity and scaffold properties post-cryopreservation. Scaffolds maintain high cell viability and metabolic activity without DMSO, demonstrating effective cryoprotection through biomaterial engineering rather than chemical cryoprotectants [59].
Cryopreservation of Scaffold-Free Pre-differentiated Tissues
Scaffold-free constructs mimic developmental processes more closely and eliminate concerns related to scaffold degradation, but require specialized cryopreservation approaches.
Experimental Protocol: Cryopreservation of gMSC1 Scaffold-Free Constructs
Materials:
- Cells: Synovial mesenchymal stem cells (sMSCs) [60]
- Culture Media: Serum-free media [60]
- Construct Formation: High-density monolayer culture forming sheet-like "gMSC1" constructs [60]
- Preservation Methods: Refrigeration vs. cryopreservation [60]
Methodology:
- Construct Fabrication: Culture sMSCs in serum-free media, seed at high density in monolayer to develop scaffold-free sheet-like constructs (gMSC1) [60].
- Pre-differentiation: Culture constructs under conditions promoting chondrogenic differentiation potential [60].
- Cryopreservation: Freeze constructs using optimized protocol (Fro-gMSC1) versus refrigeration (Ref-gMSC1) [60].
- Post-thaw Evaluation: Assess chondrogenic potential via quantitative real-time PCR and glycosaminoglycan (GAG) content quantification. Test in vivo functionality in rat cartilage defect models [60].
Table 2: Functional Assessment of Cryopreserved Scaffold-Free Constructs
| Assessment Parameter | Fresh gMSC1 | Frozen gMSC1 (Fro-gMSC1) | Refrigerated gMSC1 (Ref-gMSC1) |
|---|---|---|---|
| Chondrogenic Gene Expression | Baseline levels | Maintained expression profile | Comparable to fresh constructs [60] |
| GAG Content | Baseline production | Preserved matrix production | Similar GAG accumulation [60] |
| Cartilage Repair in Rat Model | Good integration & tissue filling | Sustained repair capacity | Functional tissue replacement [60] |
| Construct Morphology | Native structure | Comparable quality to fresh | Maintained architectural integrity [60] |
Cryopreservation of Geometrically-Defined Scaffold-Free Constructs
Tubular scaffold-free constructs designed for long bone regeneration require specialized cryopreservation approaches:
Materials:
- Cells: Human mesenchymal stem cells (hMSCs) [61]
- Morphogen Delivery: TGF-β1-presenting gelatin microspheres and BMP-2-presenting mineral-coated hydroxyapatite microparticles [61]
- Construct Design: Engineered hMSC tubes created via self-assembled ring fusion [61]
Methodology:
- Construct Fabrication: Seed suspension of hMSCs and morphogen-presenting microparticles in custom agarose molds to facilitate tissue ring self-assembly by day 2 [61].
- Tubular Construct Formation: Create tubular hMSC constructs by placing several ring building blocks in direct contact on horizontal glass tubes for additional 6 days to facilitate tissue fusion [61].
- In vitro Maturation: Culture constructs for 2 weeks in basal medium followed by 3 weeks in osteogenic medium [61].
- Cryopreservation and Recovery: Apply optimized cryopreservation protocol, thaw, and implant in vivo for functional assessment [61].
Key Findings: Localized morphogen presentation (TGF-β1 + BMP-2) stimulates chondrogenic priming and endochondral differentiation in vitro. Cryopreserved constructs maintain capacity to form cartilage templates that undergo bony remodeling in subcutaneous environments and stimulate robust endochondral healing of critical-sized femoral segmental defects orthotopically [61].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Cryopreservation of MSC-Based Constructs
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Cryoprotectants | Prevent freezing damage | DMSO (10% in FBS) [58]; Glycerol, Ethylene glycol [30]; Hyaluronic acid (DMSO-free alternative) [59] |
| Scaffold Materials | 3D structural support | SPCL (starch-polycaprolactone blend) [58]; β-TCP (3D-printed) [62]; Alginate-nanocellulose-Ha bioink [59] |
| Cell Sources | Regenerative potential | Goat bone marrow stromal cells (GBMSCs) [58]; Synovial MSCs (sMSCs) [60]; Human MSCs (hMSCs) [61] |
| Morphogen Delivery | Guided differentiation | TGF-β1-presenting gelatin microspheres [61]; BMP-2-presenting mineral-coated hydroxyapatite microparticles [61] |
| Assessment Tools | Viability & functionality | MTS assay [58]; DNA quantification [58]; SEM, micro-CT [58]; Histology (Safranin-O, Alizarin Red) [61] |
Signaling Pathways and Experimental Workflows
Cryopreservation Workflow for Tissue-Engineered Constructs
Diagram 1: Comprehensive cryopreservation workflow for scaffold-based and scaffold-free tissue-engineered constructs, illustrating parallel pathways with common critical steps including cryoprotection, freezing, thawing, and functional assessment.
Signaling Pathways in Pre-differentiated Constructs
Diagram 2: Key signaling pathways activated by morphogen presentation in pre-differentiated tissue constructs, showing how TGF-β1 and BMP-2 activate SMAD3 and SMAD5 pathways leading to chondrogenic priming, osteogenic commitment, and ultimately endochondral ossification for functional bone regeneration.
The application workflows detailed in this document provide standardized methodologies for cryopreserving two principal classes of MSC-based tissue-engineered structures: scaffold-based constructs and scaffold-free pre-differentiated tissues. The quantitative data demonstrates that both approaches can maintain viability, functionality, and structural integrity post-preservation when appropriate protocols are followed. Key considerations include cryoprotectant selection (with movement toward DMSO-free alternatives), scaffold architecture preservation, and adequate post-thaw recovery periods. These protocols enable the creation of biobanks for "off-the-shelf" availability of tissue-engineered products, addressing a critical bottleneck in clinical translation of regenerative therapies.
Solving Critical Challenges: CPA Toxicity, Ice Damage, and Functional Recovery
The cryopreservation of mesenchymal stem cell (MSC)-based tissue-engineered structures represents a critical technological bridge between laboratory research and clinical application. Achieving reliable, long-term preservation while maintaining cellular viability, differentiation potential, and secretory function is paramount for the advancement of regenerative medicine. For decades, dimethyl sulfoxide (DMSO) has served as the cornerstone cryoprotectant for cellular systems, leveraging its ability to penetrate cells and suppress ice crystal formation through hydrogen bonding with water molecules [63]. However, its application in advanced tissue-engineered constructs presents significant challenges. DMSO toxicity is not merely a theoretical concern but a practical limitation that can compromise the therapeutic efficacy of MSC-based products [64] [65]. This toxicity manifests through multiple mechanisms, including induction of cellular dehydration, disruption of membrane integrity, alteration of epigenetic landscapes, and at high concentrations, direct activation of apoptotic pathways [64] [63]. In the context of tissue-engineered structures, these effects are amplified due to the complex cellular architecture and presence of extracellular matrix (ECM) components.
The pursuit of DMSO-free or DMSO-reduced cryopreservation strategies is therefore not merely an academic exercise but a necessary evolution toward safer, more effective clinical applications. This application note synthesizes current research and provides detailed protocols for mitigating cryoprotectant toxicity while maintaining the functional integrity of MSC-based tissue-engineered structures, with particular emphasis on the preservation of self-assembled cellular constructs that include endogenous extracellular matrix.
The Challenge of DMSO Toxicity in MSC Constructs
DMSO's efficacy as a cryoprotectant is inextricably linked to its toxicity profile, creating a fundamental challenge for cryopreservation protocol design. The mechanisms of DMSO-induced damage are multifaceted and particularly problematic for complex MSC constructs.
Biochemical and Structural Impacts
At the cellular level, DMSO exposure can induce drastic changes in cellular processes and the epigenetic landscape in vitro [63]. Even at concentrations as low as 0.1%, DMSO can interact with various cellular and signaling pathways, potentially altering the differentiation potential and immunomodulatory properties of MSCs—the very attributes that make them therapeutically valuable [66]. Furthermore, DMSO has a synergistic damaging effect when combined with other stress factors; for instance, it has been shown to act synergistically with vanadium to delay zebrafish embryo development and cause pericardial edema [63].
In tissue-engineered constructs such as MSC/extracellular matrix complexes (C-MSCs), the problem extends beyond individual cells to the structural integrity of the entire construct. The extracellular matrix provides not only physical scaffolding but also critical biochemical and biomechanical cues that direct cellular function. DMSO's solvent properties can disrupt matrix architecture and compromise cell-matrix interactions essential for post-thaw recovery and functionality [67].
Clinical Implications
From a clinical perspective, the administration of DMSO-preserved cell products carries measurable risks. Nearly 100% of bone marrow transplant recipients receiving DMSO-cryopreserved cells experience side effects or serious complications during infusion [66]. These range from transient symptoms like nausea and vomiting to more severe cardiovascular, respiratory, and neurological toxicities [66] [65]. While recent analyses suggest that the DMSO doses delivered via MSC products may be 2.5–30 times lower than those accepted in hematopoietic stem cell transplantation [65], the risk profile remains significant, particularly for fragile patient populations or with repeated administrations.
Table 1: Documented Toxic Effects of DMSO on MSCs and Related Systems
| Toxicity Type | Observed Effects | Concentration Range | Reference |
|---|---|---|---|
| Cellular Toxicity | Reduced clonogenic potential, altered epigenetics, actin filament disruption | 7.5-10% (v/v) | [64] [63] |
| Structural Damage | Membrane undulations, cellular swelling, ECM disruption | 10-20% (v/v) | [64] [67] |
| Functional Impairment | Impaired osteoclast differentiation, reduced mitochondrial function | >0.1% (v/v) | [66] [63] |
| Clinical Effects | Nausea, vomiting, hypotension, cardiovascular instability | Dose-dependent | [66] [65] |
Emerging DMSO Alternatives and Reduction Strategies
The development of DMSO-free cryopreservation media represents the most direct approach to eliminating DMSO-associated toxicity. However, a spectrum of strategies exists, ranging from complete replacement to significant reduction through combination with other cryoprotective agents.
Complete DMSO Replacement Technologies
Several innovative approaches have emerged as complete DMSO replacements, demonstrating particular promise for MSC preservation:
Bioinspired Cryoprotectants: XT-Thrive represents a class of fully synthetic, biomimetic cryoprotectants inspired by natural antifreeze proteins. These formulations control ice formation through ice-interactive polymers and are protein-free, serum-free, and chemically defined [66]. In comparative studies with bone marrow-derived MSCs, XT-Thrive maintained approximately 30% higher viability than DMSO-containing controls after 24-hour incubation at room temperature (∼93% vs. ∼61% viability) and showed superior post-thaw recovery (∼87% vs. ∼63% viability) [68]. Furthermore, MSCs preserved in XT-Thrive exhibited a 2.5-fold expansion in serum-containing media and a 2-fold expansion in serum-free microcarrier cultures, outperforming DMSO-preserved cells which showed only 0.9-fold expansion under serum-free conditions [68].
Bambanker DMSO-Free: This serum-free cryopreservation medium provides a safe and reliable alternative to traditional DMSO-based media, offering consistent, high-performance preservation for a wide range of cell types, including primary cells and stem cells [69]. By eliminating both DMSO and serum, Bambanker helps preserve cell viability and integrity without the cytotoxicity risks or variability associated with other formulations, making it particularly valuable for applications in regenerative medicine and cell therapies [69].
DMSO Reduction Through Combination Strategies
Partial replacement of DMSO with less toxic cryoprotectants can significantly reduce overall toxicity while maintaining adequate cryoprotection:
Trehalose-Based Formulations: The disaccharide trehalose has emerged as a particularly effective co-cryoprotectant. As a non-reducing sugar, trehalose stabilizes membranes and proteins in the dry state through water replacement mechanisms and vitrification. Studies with umbilical cord blood stem cells demonstrated that a low concentration of DMSO (2.5% v/v) combined with 30 mmol/L trehalose resulted in higher post-thaw cell viability, increased colony-forming units (CFUs), and reduced apoptosis compared to standard 10% DMSO formulations [70]. Trehalose exerts a similar cryoprotective potential for hematopoietic progenitor and stem cells as larger impermeant sugars and could possibly replace DMSO at least in part as a cryoprotectant [70].
Multi-Agent Cryoprotectant Cocktails: Advanced formulations combining penetrating and non-penetrating cryoprotectants show promise for complex tissue constructs. One effective approach utilizes 3% trehalose + 5% dextran 40 + 4% polyethylene glycol, which demonstrated approximately 95% viability and recovery for adipose tissue-derived MSCs [65]. Similarly, a formulation containing 150 mM sucrose + 300 mM ethylene glycol + 30 mM alanine + 0.5 mM taurine + 0.02% ectoine achieved 96% viability and 103% recovery in embryonic stem cell-derived MSCs [65].
Table 2: Performance Comparison of DMSO Reduction and Replacement Strategies
| Cryoprotectant Strategy | Composition | Post-Thaw Viability | Functional Recovery | Advantages |
|---|---|---|---|---|
| Traditional Control | 10% DMSO + serum | ~70-80% | Baseline | Established protocol |
| Trehalose Combination | 2.5% DMSO + 30 mmol/L trehalose | Significantly higher than 10% DMSO | Higher CFUs, lower apoptosis | Reduced DMSO load, minimal toxicity |
| Bioinspired Synthetic | XT-Thrive (fully synthetic) | ~87-93% | 2.5-fold expansion in serum, 2-fold in serum-free | DMSO-free, defined composition, room temperature stable |
| Polymer-Sugar Cocktail | 3% trehalose + 5% dextran 40 + 4% PEG | ~95% | ~95% recovery | DMSO-free, excellent for sensitive cells |
| Amino Acid Enhanced | 150 mM sucrose + 300 mM EG + 30 mM alanine + 0.5 mM taurine | 96% | 103% recovery | DMSO-free, metabolic support |
Application-Oriented Protocols
Protocol 1: Cryopreservation of MSC/ECM Complexes (C-MSCs) Using DMSO-Reduced Media
Background: Three-dimensional cultured clumps of MSC/extracellular matrix complexes (C-MSCs) consist of cells and self-produced ECM, which can be grafted into defect sites without artificial scaffolds to induce bone regeneration [67]. Preserving the structural integrity of these complexes during cryopreservation is essential for maintaining their regenerative capacity.
Materials:
- C-MSCs (0.8-1.2 mm diameter)
- Cryoprotectant solution: 5% DMSO + 150 mM sucrose + 10% polyethylene glycol (MW 8000) in basal medium
- Controlled-rate freezer
- Cryovials
- Water bath (37°C)
Procedure:
- Pre-equilibration: Gradually introduce C-MSCs to cryoprotectant solution using a stepwise addition method (25%, 50%, 75%, 100% over 30 minutes) at 4°C to minimize osmotic shock.
- Loading: Transfer individual C-MSCs to cryovials containing 500 μL cryoprotectant solution.
- Freezing: Place samples in a controlled-rate freezer with the following protocol:
- Start temperature: 4°C
- Cool at -1°C/min to -5°C
- Soak for 5 minutes to allow for heat of fusion dissipation
- Cool at -1°C/min to -40°C
- Cool at -10°C/min to -80°C
- Storage: Transfer samples to liquid nitrogen for long-term storage.
- Thawing: Rapidly warm samples in a 37°C water bath until only a small ice crystal remains.
- CPA Removal: Gently transfer C-MSCs to culture medium and allow natural settling (avoid centrifugation) to preserve 3D structure.
- Assessment: Evaluate viability using calcein-AM/ethidium homodimer staining and osteogenic potential through alkaline phosphatase activity and mineralization assays.
Validation: This protocol has demonstrated maintained 3D structure, high cell viability, and preserved osteogenic differentiation capacity in cryopreserved C-MSCs, with successful bone regeneration in rat calvarial defect models [67].
Protocol 2: Complete DMSO-Free Cryopreservation of MSCs for Microcarrier-Based Bioprocessing
Background: For scalable manufacturing of MSC-based therapies, integration with microcarrier-based bioreactor systems requires cryopreservation protocols that maintain high recovery and expansion potential under serum-free conditions.
Materials:
- XT-Thrive cryopreservation medium (X-Therma Inc.) or equivalent DMSO-free medium
- Bone marrow-derived MSCs at passage 3-5
- Serum-free expansion medium
- Cryovials
- CoolCell or similar freezing container
- Microcarriers (e.g., Cytodex 3) for post-thaw assessment
Procedure:
- Harvesting: Detach MSCs using enzyme-free dissociation buffer to maintain surface marker integrity.
- Preparation: Centrifuge cells at 300 × g for 5 minutes and resuspend in XT-Thrive at 1 × 10^6 cells/mL.
- Incubation: Hold cell suspension in cryoprotectant for 10 minutes at room temperature before freezing.
- Freezing: Aliquot 1 mL cell suspension into cryovials and place in CoolCell freezing container.
- Freezing Rate: Transfer container to -80°C freezer for 24 hours (achieving approximately -1°C/min cooling rate).
- Storage: Transfer vials to liquid nitrogen after 24 hours.
- Thawing: Rapidly warm in 37°C water bath with gentle agitation.
- Direct Culture: Dilute thawed cell suspension 1:10 with pre-warmed serum-free medium and seed directly onto pre-hydrated microcarriers in spinner flasks.
- Assessment: Monitor cell viability at 6 hours post-thaw and cell expansion over 6 days.
Performance Metrics: This protocol typically yields >87% post-thaw viability and approximately 2-fold expansion in serum-free microcarrier culture over 6 days, outperforming 10% DMSO controls which show <63% viability and <1-fold expansion under the same conditions [68].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for DMSO-Free Cryopreservation Research
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Penetrating CPAs | Ethylene glycol, propylene glycol, glycerol | Penetrate cell membrane, suppress intracellular ice formation | Often combined with non-penetrating agents; glycerol has lower toxicity but worse cryopreservation effect compared to DMSO [27] |
| Non-Penetrating CPAs | Trehalose, sucrose, dextran-40, raffinose | Stabilize membranes extracellularly, modulate ice formation | Trehalose particularly effective; can be introduced intracellularly via electroporation or nanoparticles [65] |
| Ice-Binding Polymers | XT-Thrive (biomimetic polymers), polyvinyl pyrrolidone | Control ice crystal formation and growth through ice recrystallization inhibition | Biomimetic approach inspired by antifreeze proteins; reduces need for high CPA concentrations [66] |
| Membrane Stabilizers | Cholesterol-loaded cyclodextrin, hydroxyethyl starch | Strengthen membrane integrity against freezing-induced damage | Particularly important for sensitive cell types and complex constructs |
| Antioxidants | Taurine, alanine, ectoine | Scavenge reactive oxygen species generated during freeze-thaw cycles | Address oxidative stress component of cryoinjury; especially valuable for tissue constructs [65] |
| Commercial Media | Bambanker DMSO-Free, STEM-CELLBANKER DMSO free | Complete, optimized formulations | Provide standardized, ready-to-use solutions; eliminate formulation variability |
Strategic Workflow for Cryoprotectant Optimization
The following diagram illustrates a systematic approach to developing and optimizing cryopreservation protocols for MSC-based tissue-engineered structures, incorporating critical decision points and evaluation metrics.
Diagram 1: Cryopreservation Protocol Development Workflow - This workflow outlines a systematic approach to optimizing cryopreservation protocols for MSC-based constructs, emphasizing iterative refinement based on comprehensive post-thaw assessment.
Mechanisms of Cryoprotection and Toxicity Mitigation
Understanding the fundamental mechanisms through which alternative cryoprotectants operate is essential for their rational implementation in research and development settings.
Diagram 2: Cryoprotection Mechanisms and Toxicity Mitigation - This diagram illustrates the primary mechanisms of cryodamage (left), how alternative cryoprotectants counter these mechanisms (center), and the specific DMSO toxicity pathways that are mitigated by alternative approaches (right).
The movement toward DMSO-free and DMSO-reduced cryopreservation strategies for MSC-based tissue-engineered structures represents a critical advancement in regenerative medicine. The protocols and data presented herein demonstrate that effective cryopreservation can be achieved while significantly reducing or eliminating DMSO-associated toxicity. The emergence of biomimetic cryoprotectants like XT-Thrive and optimized combination approaches using trehalose with minimal DMSO provide researchers with viable alternatives that maintain post-thaw viability, functionality, and structural integrity.
Future developments in this field will likely focus on several key areas: First, the customization of cryoprotectant formulations for specific MSC tissue sources (bone marrow, adipose tissue, umbilical cord) and construct types (2D, 3D, ECM-rich). Second, the integration of advanced materials such as nanoparticles for intracellular delivery of non-penetrating cryoprotectants and ice-binding polymers for enhanced ice management. Third, the development of closed-system, automated cryopreservation workflows to enhance reproducibility and compliance with good manufacturing practices for clinical applications.
As the field progresses, the optimal cryopreservation strategy will continue to balance the competing demands of cryoprotection efficacy, toxicity minimization, technical feasibility, and regulatory compliance. The protocols and data presented in this application note provide a foundation for researchers to make informed decisions in developing cryopreservation strategies tailored to their specific MSC-based tissue engineering applications.
Optimizing Cooling Rates and CPA Concentrations to Minimize Intracellular Ice
In the field of regenerative medicine, mesenchymal stromal cell (MSC)-based tissue-engineered structures represent a promising frontier for treating a wide range of conditions [3]. The clinical translation of these advanced therapies is critically dependent on effective cryopreservation protocols that ensure product viability, functionality, and off-the-shelf availability [31] [33]. A central challenge in cryopreservation is the prevention of intracellular ice formation (IIF), a lethal event that disrupts cellular structures and functions [71] [72] [73].
This application note provides a detailed framework for optimizing cooling rates and cryoprotectant agent (CPA) concentrations to minimize IIF in MSC-based tissue constructs. The guidance is grounded in the fundamental two-factor theory of cryoinjury, which posits that damage results from the combined effects of intracellular ice formation at high cooling rates and solute-induced "solution effects" or osmotic shock at low cooling rates [73]. By navigating this balance, researchers can develop robust preservation protocols essential for the commercialization and clinical success of tissue-engineered products.
Fundamental Principles of Ice Formation and Cell Injury
During cryopreservation, the behavior of water inside and outside the cell is the primary determinant of survival. When cooling rates are too slow, prolonged exposure to hypertonic extracellular solutions causes severe cell dehydration and shrinkage, leading to solute damage [73]. Conversely, excessively rapid cooling does not permit sufficient water to exit the cell, resulting in supercooling and eventual lethal intracellular ice formation [72] [73].
The role of CPAs is to modulate these physical processes. Penetrating CPAs, such as dimethyl sulfoxide (DMSO) and ethylene glycol, permeate the cell and reduce the amount of "free" water available for ice crystallization, thereby colligatively depressing the ice nucleation temperature [72] [33]. Non-penetrating CPAs (e.g., sucrose) remain extracellular and promote gentle osmotic dehydration before freezing [72]. The choice of cooling rate and CPA concentration is therefore interdependent; higher CPA concentrations generally allow for slower cooling by enhancing glass-forming tendencies and reducing the required dehydration [71] [73].
For complex 3D biofabricated constructs, additional challenges arise from mass transfer limitations. CPA diffusion and thermal gradients within engineered tissues can be non-uniform, creating zones with different susceptibilities to ice formation [31]. Consequently, protocols optimized for single-cell suspensions may require significant adaptation for tissue constructs.
Experimental Approaches and Protocols
Determination of Intracellular Ice Formation
Synchrotron-Based X-Ray Diffraction Protocol The gold standard for direct detection and quantification of intracellular ice uses synchrotron x-ray diffraction, which can detect ice volume fractions below 1% [72].
- Sample Preparation: Bovine oocytes (as a model for large mammalian cells) are equilibrated in CPA solutions and placed on Cryotop supports or crystallography loops [72].
- Cooling Procedure: Cool samples at controlled rates (e.g., ~30,000 °C/min for conventional rates or ~600,000 °C/min for ultra-rapid cooling) using a liquid-nitrogen-based cryocrystallography instrument [72].
- Data Collection: At a stable cryogenic temperature (e.g., -173 °C), expose the sample to a synchrotron x-ray beam. Collect 2D diffraction patterns to identify the structure and quantity of ice present (e.g., amorphous ice, stacking-disordered ice Isd, or hexagonal ice Ih) [72].
- Data Analysis: Azimuthally integrate diffraction patterns to generate 1D intensity vs. resolution plots. Fit data to models of stacking-disordered ice to determine the hexagonal fraction (Φh) and quantify crystalline ice content [72].
Cryomicroscopy Protocol for IIF Observation A more accessible method involves using a cryomicroscope to visually detect the sudden darkening or "flashing" of the cytoplasm that signals IIF [71].
- System Setup: Utilize a thermoelectric cooling (TEC) cryomicroscope system capable of precise temperature control (accuracy within ±0.5 °C) and direct visualization [71].
- Experimental Run: Place cells in a CPA solution on the cold stage. Cool at a defined linear rate (e.g., 1.5, 3, 7, or 12 °C/min) while recording video [71].
- IIF Temperature Recording: For each cell, note the temperature at which the sudden optical change occurs. Compile data from 20-30 cells per condition to generate a cumulative probability distribution of IIF temperatures [71].
- Data Fitting: Fit the IIF temperature distribution to a probabilistic model (e.g., Weibull distribution) to characterize the IIF behavior under different cooling rates and CPA concentrations [71].
Systematic Optimization of Cooling Rate and CPA Concentration
The following workflow outlines a step-by-step methodology for determining the optimal parameters to suppress IIF for a given MSC-based product. This process integrates the principles of the two-factor theory with empirical testing.
Quantitative Data for Parameter Optimization
Table 1: Effect of Cooling Rate and DMSO Concentration on Intracellular Ice Formation in a Model System (small abalone eggs). Data adapted from Yang et al. (2013) [71].
| DMSO Concentration (M) | Cooling Rate (°C/min) | IIF Suppression | Post-Thaw Osmotic Activity | Notes |
|---|---|---|---|---|
| 2.0 | 1.5 | Well-suppressed | 48.8% | Feasible protocol; low toxicity |
| 2.0 | 12 | Partial | Lower than 1.5°C/min | Higher ice risk |
| 2.5 | 3 | Well-suppressed | Data not specified | Increased CPA toxicity |
| 3.0 | 7 | Well-suppressed | Data not specified | Higher CPA toxicity |
| 4.0 | 12 | Well-suppressed | Data not specified | Highest CPA toxicity |
Table 2: General Guidelines for Cryopreservation of Different Biospecimen Types. Synthesized from multiple sources [31] [72] [73].
| Biospecimen Type | Typical CPA | Typical Concentration Range | Typical Cooling Rate | Critical Consideration |
|---|---|---|---|---|
| Single Cells (e.g., MSCs) | DMSO | 5-10% (v/v) | -1 °C/min [74] | Optimize for cell-specific membrane permeability |
| Oocytes/Embryos | Ethylene Glycol + Sucrose | ~6-8 M total (Vitrification) | >20,000 °C/min (Vitrification) [72] | Ultra-rapid warming is critical to prevent devitrification |
| 3D Biofabricated Constructs | DMSO + Trehalose (DMSO-free options) | 5-10% (v/v) + 0.1-0.2 M | -1 to -5 °C/min (Slow freezing) [31] | CPA diffusion limitation; use of cryoprotective biomaterials (e.g., HA) |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Cryopreservation Optimization.
| Reagent/Material | Function/Purpose | Example Use Case |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating CPA; reduces ice crystal formation by binding water molecules. | Standard CPA for slow freezing of MSC suspensions [31] [33]. |
| Ethylene Glycol | Penetrating CPA; lower toxicity alternative for some sensitive cells. | Common component in vitrification solutions for oocytes/embryos [72]. |
| Sucrose | Non-penetrating CPA; induces osmotic dehydration pre-freezing. | Used in combination with penetrating CPAs in vitrification solutions [72]. |
| Hyaluronic Acid (HA) Hydrogels | Cryoprotective biomaterial; provides structural support and uniform CPA diffusion in 3D constructs. | MeHA matrices for cryopreserving MSC-laden 3D constructs [31]. |
| Polyvinyl Alcohol (PVA) | Synthetic polymer with ice recrystallization inhibition (IRI) properties. | Additive to CPA solutions to control ice crystal growth during thawing [31]. |
| Controlled-Rate Freezer (CRF) | Instrument for precise, programmable control of cooling rates. | Essential for implementing optimized slow-freezing protocols in GMP manufacturing [74]. |
| Cryomicroscope | System for direct visualization of ice formation in cells during cooling/warming. | Empirical determination of IIF temperatures for different cooling rates [71]. |
Advanced Strategies for Tissue-Engineered Constructs
The transition from 2D cell cultures to 3D biofabricated structures introduces significant complexity for IIF control. Key advanced strategies include:
Biomaterial-Assisted Cryopreservation: Natural polymers like methacrylated hyaluronic acid (MeHA) can create hydrogels that support homogeneous CPA diffusion, leading to post-thaw MSC viabilities of 40-60% and maintained differentiation potential [31]. These materials can act as intrinsic cryoprotectants by modulating ice crystal growth and mitigating osmotic stress.
DMSO-Free and Low-CPA Formulations: To mitigate CPA toxicity, research focuses on combinations of low concentrations of penetrating CPAs with non-penetrating agents and IRI polymers. For example, combining 3–5% DMSO with 0.1–0.2% high-molecular-weight hyaluronic acid (HMW-HA) has improved MSC survival and osteo/chondrogenic capacity [31].
Vitrification of Constructs: For particularly sensitive or complex tissues, vitrification—the rapid cooling of a high-CPA concentration solution to form a glassy, non-crystalline state—can completely avoid IIF [73]. The primary challenge is scaling this technique to larger 3D structures while avoiding the toxic effects of high CPA concentrations and ensuring ultra-rapid and uniform warming to prevent "devitrification" (ice formation during warming) [31] [73].
The successful cryopreservation of MSC-based tissue-engineered structures is a critical enabler for their clinical and commercial viability. By systematically optimizing cooling rates and CPA concentrations, researchers can effectively minimize the damaging effects of intracellular ice formation. The protocols and data provided here serve as a foundational guide for this optimization process.
Future progress will likely rely on the continued development of advanced biomaterials with integrated cryoprotective functions and the refinement of vitrification protocols for larger, more complex tissues. By prioritizing the control of ice formation, the field of regenerative medicine can overcome a significant bottleneck, paving the way for the widespread availability of off-the-shelf, cell-based therapeutic products.
Within regenerative medicine and advanced drug development, Mesenchymal Stem Cells (MSCs) are a cornerstone for tissue-engineered structures and cell-based therapies. A significant barrier to their clinical translation is the substantial cell death and functional loss that can occur during the critical cryopreservation and post-thaw recovery processes. Inconsistent post-thaw outcomes directly compromise the therapeutic dose, efficacy, and standardization required for robust clinical applications and commercial products. This Application Note synthesizes current research to provide detailed, actionable protocols aimed at mitigating post-thaw cell loss, thereby enhancing the viability and recovery yield of MSC-based products for the research and development community.
Understanding and Quantifying Post-Thaw Cell Loss
The process of thawing cryopreserved MSCs introduces multiple stressors that can lead to significant cell loss. A primary cause is the osmotic shock experienced during the removal of cryoprotective agents (CPAs) like Dimethyl Sulfoxide (DMSO). Rinsing cells too rapidly in hypotonic or protein-free solutions creates a massive osmotic imbalance, causing excessive water influx and cell lysis [4]. Furthermore, the physical formation of ice crystals during suboptimal freezing can cause mechanical damage to cell membranes and intracellular structures [4]. Recent studies have quantified the impact of these factors, providing a baseline for improvement.
Table 1: Quantified Impact of Common Thawing and Reconstitution Practices on MSC Yield
| Parameter Tested | Suboptimal Condition | Optimal Condition | Impact on Cell Loss/Viability | Source |
|---|---|---|---|---|
| Reconstitution Solution | Protein-free PBS or saline | Saline with 2% Human Serum Albumin (HSA) | >40% cell loss in protein-free vs. minimal loss with HSA | [75] |
| Post-Thaw Cell Concentration | < 1.0 x 10^5 cells/mL | 5.0 x 10^6 cells/mL | >40% instant cell loss at low concentration | [75] |
| Post-Thaw Storage Viability | Stored in PBS at room temperature | Stored in saline at room temperature | >40% cell loss after 1 hour in PBS vs. >90% viability for 4+ hours in saline | [75] |
| Post-Thaw Viability (Clinical Meta-Analysis) | ≤ 80% viability | > 80% viability | LVEF improvement of 3.44% with >80% viability vs. no significant improvement with lower viability | [76] |
| Cold-Chain Handling | 400 manual temperature cycles | 400 automatic cycles (< -150°C) | Viability: 76.9% vs. 94.2% | [77] |
Optimized Experimental Protocols
The following protocols are compiled from recent studies that successfully minimized post-thaw cell loss and maintained MSC functionality.
Protocol: Optimized Thawing and Reconstitution for Clinical Formulation
This protocol, adapted from Roost Aabling et al. (2023), addresses the critical thawing and dilution phases to maximize viable cell yield [75].
Key Reagents:
- Thawing/Reconstitution Medium: Isotonic saline (0.9% NaCl) supplemented with 2% clinical-grade Human Serum Albumin (HSA).
- Wash Medium: Alpha-MEM or DPBS supplemented with 2% HSA or 10% FBS.
Procedure:
- Rapid Thawing: Transfer the cryovial from liquid nitrogen storage to a 37°C water bath. Gently agitate until only a small ice crystal remains.
- Immediate Dilution: Wipe the vial with 70% ethanol. Using a pipette, gently transfer the cell suspension to a 15 mL conical tube containing a pre-warmed volume of Thawing/Reconstitution Medium that is at least 10x the volume of the cryopreservation solution.
- Controlled Centrifugation: Centrifuge the cell suspension at 300-400 x g for 5 minutes to pellet the cells and remove the CPA-containing supernatant.
- Gentle Resuspension: Carefully decant the supernatant. Resuspend the cell pellet in a small volume of Wash Medium or the final administration solution. Crucially, the final cell concentration should be maintained at or above 5 x 10^6 cells/mL to prevent dilution-induced cell death.
- Post-Thaw Holding: If immediate use is not possible, cells resuspended in the optimized saline/HSA solution can be held at room temperature for up to 4 hours without significant loss of viability or yield.
Protocol: Short-Term Cryopreservation of Bone Marrow Aspirate Concentrate (BMAC)
This protocol demonstrates that functional MSCs within a complex biologic product can be preserved with minimal loss, enabling a "single-harvest, multiple-injection" strategy [29].
Key Reagents:
- Cryopreservation Medium: 10% DMSO in 90% autologous plasma (from the BMAC processing).
- Equipment: Controlled-rate freezing container (e.g., "Mr. Frosty").
Procedure:
- BMAC Processing: Concentrate bone marrow aspirate using standard density gradient centrifugation or automated systems to isolate the mononuclear cell fraction.
- CPA Addition: Resuspend the BMAC cell pellet in cryopreservation medium (10% DMSO/90% autologous plasma) at a density of 1-2 million cells/mL.
- Controlled-Rate Freezing: Place the cryovials in a pre-cooled controlled-rate freezing container. Transfer the container to a -80°C freezer for a minimum of 24 hours. The freezing rate should be approximately -1°C/min.
- Long-Term Storage: After 24 hours, promptly transfer vials to liquid nitrogen for long-term storage.
- Thawing and Washing: Rapidly thaw in a 37°C water bath. Dilute the thawed product in pre-warmed culture medium and centrifuge at 300 x g for 5 minutes to remove DMSO before use.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Optimized MSC Cryopreservation and Thawing
| Reagent / Solution | Function & Rationale | Application Notes |
|---|---|---|
| Human Serum Albumin (HSA) | Prevents cell adhesion loss and membrane damage during thawing and dilution; provides oncotic pressure to mitigate osmotic shock. | Use at 2% in isotonic solutions. A critical additive for protein-free formulation. |
| Autologous Plasma | Serves as a autologous protein source in cryomedium; may enhance post-thaw recovery by providing native growth factors. | Used in BMAC cryopreservation at 90% ratio with 10% DMSO [29]. |
| DMSO (Dimethyl Sulfoxide) | Penetrating cryoprotectant; reduces ice crystal formation by binding water molecules. The current gold-standard despite toxicity concerns. | Standard concentration is 10%. DMSO-associated toxicity is dose-dependent, and its removal post-thaw is critical [65] [4]. |
| Isotonic Saline (0.9% NaCl) | A simple, clinically compatible vehicle for post-thaw resuspension and storage. Superior to PBS for maintaining short-term viability. | Must be supplemented with a protein source like HSA for optimal recovery [75]. |
| Controlled-Rate Freezer / Mr. Frosty | Ensures a consistent, slow cooling rate (~-1°C/min), which is crucial for cell dehydration and minimizing intracellular ice formation. | Essential for reproducible slow-freezing protocols [29] [4]. |
Workflow Visualization: Optimized Post-Thaw Processing
The following diagram outlines the critical decision points and steps in the optimized post-thaw workflow to maximize MSC recovery, integrating the key protocols discussed above.
Achieving high viability and recovery yield for MSCs post-thaw is not a matter of a single intervention but requires a holistic and optimized approach across the entire process. As demonstrated, key strategies include the use of protein-supplemented solutions like saline with HSA during thawing and reconstitution, maintaining high cell concentrations to prevent dilution-induced apoptosis, employing controlled-rate freezing, and minimizing temperature fluctuations during cold-chain storage. By implementing the detailed protocols and best practices outlined in this Application Note, researchers and drug developers can significantly enhance the reliability, efficacy, and clinical translatability of their MSC-based tissue-engineered structures and therapies.
Within cryopreservation research for MSC-based tissue-engineered structures, preventing contamination is a critical determinant of experimental success and clinical translation. Contamination compromises not only cell viability but also the structural and functional integrity of complex three-dimensional (3D) constructs, leading to unreliable data and potential safety hazards [78] [79]. The process from liquid nitrogen (LN2) storage to final thawing presents multiple contamination risk points, including environmental microbial ingress, cross-contamination between samples, and chemical toxicity from cryoprotective agents (CPAs) [4] [80]. Adhering to rigorous aseptic technique throughout this workflow is therefore paramount to preserve the phenotype, differentiation potential, and therapeutic efficacy of mesenchymal stem cells (MSCs) and their engineered tissues [4] [81]. This application note provides detailed protocols and evidence-based strategies to mitigate these risks, ensuring the integrity of your cryopreserved samples.
Fundamental Aseptic Principles and Contamination Risks
Aseptic technique encompasses all procedures designed to prevent the introduction of contaminating microorganisms (bacteria, fungi, mycoplasma) and cross-contaminating particles into sterile samples. In the context of MSC cryopreservation, lapses in aseptic technique are a primary source of contamination, often stemming from improper glove use, talking over open cultures, or working outside a biological safety cabinet (BSC) [78]. The consequences are severe, leading to compromised experimental data, wasted resources, and a loss of scientific credibility [78].
The table below summarizes the primary sources and impacts of contamination relevant to MSC-based tissue engineering.
Table 1: Key Contamination Sources and Their Consequences in MSC Cryopreservation
| Contamination Source | Specific Examples | Impact on MSCs & Engineered Tissues |
|---|---|---|
| Environmental | Airborne dust/aerosols, unclean HVAC/HEPA systems, high-traffic areas [78] | Microbial contamination of cell cultures; altered gene expression and differentiation potential [78] [79] |
| Human/Procedural | Poor pipetting technique, reusing pipette tips, mislabeling tubes, wearing same PPE between cell lines [78] | Cross-contamination between samples; loss of sample traceability; introduction of mycoplasma [78] [80] |
| Equipment/Supplies | Dirty glassware with residual detergent, improperly sterilized tools, non-sterile consumables [78] | Skewed assay results from chemical residues; introduction of biological contaminants [78] |
| CPA Handling | Use of cytotoxic CPAs like DMSO, improper addition/removal causing osmotic stress [4] [81] | Reduced post-thaw viability and recovery; impaired proliferation and differentiation potential; triggering of allergic responses in clinical applications [4] |
Aseptic Workflow: From LN2 Storage to Thawing
Pre-Thaw Preparation and Safe LN2 Handling
A meticulously prepared workspace is the foundation of aseptic practice.
- Personal Protective Equipment (PPE): Always wear appropriate PPE, including gloves, a lab coat, goggles, and a mask. When retrieving vials from LN2, specific cryogenic gloves, a face shield, and an apron are mandatory to protect against extreme temperatures and potential vial explosions [82].
- Workspace Preparation: Perform all thawing procedures within a Class II Biological Safety Cabinet (BSC) or laminar flow hood, which should be turned on and UV-sterilized (if applicable) prior to use. Ensure the workspace is uncluttered and clean. Disinfect all surfaces with 70% ethanol and place absorbent material on the work surface to contain any potential spills [82]. Pre-place all necessary equipment within the BSC, including sterile centrifuge tubes, pre-warmed culture media, pipettes, and a waste container [82].
- LN2 Storage and Vial Retrieval: Store cryopreserved MSC constructs in the vapor phase of LN2 (typically -150°C to -196°C) rather than the liquid phase to minimize the risk of microbial contamination, particularly from Acinetobacter spp., which can penetrate vial seals in liquid nitrogen [80]. Use tongs or forceps to retrieve vials, and always wear a face shield. Never seal cryovials tightly when plunging into LN2, as trapped liquid can expand and cause a dangerous explosion [82].
Controlled and Aseptic Thawing Protocol
Rapid thawing is critical for high cell viability, but the process must be performed aseptically.
Table 2: Step-by-Step Aseptic Thawing Protocol for Cryopreserved MSCs
| Step | Procedure | Aseptic Rationale & Key Considerations |
|---|---|---|
| 1. Retrieval | Quickly retrieve the vial from LN2 storage. Visually inspect for cracks or defects. | Minimize temperature fluctuations and ice crystal regrowth. Do not submerge the vial cap in the water bath. |
| 2. Thawing | Immediately place the vial in a pre-warmed 37°C water bath or a validated controlled-thawing device. Gently agitate until only a small ice crystal remains [4] [74]. | A water bath is a contamination risk; ensure it is clean and consider using a bag to isolate the vial. Controlled-thawing devices are GMP-compliant and preferred for clinical work [74]. |
| 3. Decontamination | Wipe the exterior of the vial thoroughly with 70% ethanol before introducing it into the BSC. | Prevents introduction of external contaminants from the water bath into the sterile work area [78]. |
| 4. Handling in BSC | Carefully transfer the vial into the pre-prepared BSC. | Limit direct handling and exposure to the open environment. |
| 5. Dilution | Gently transfer the thawed cell suspension to a sterile conical tube. Slowly add pre-warmed culture medium or a specialized CPA dilution buffer (e.g., CellShield MSC Buffer) drop-wise while gently swirling [4] [81]. | Slow dilution minimizes osmotic shock and cell lysis caused by the sudden change in CPA concentration [4]. |
| 6. CPA Removal | Centrifuge the cell suspension at a recommended speed and time (e.g., 300-400 x g for 5-10 minutes). Aspirate and discard the supernatant containing the CPA. | Effectively removes cytotoxic CPAs like DMSO. Centrifugation must be gentle to avoid damaging the freshly thawed cells [4]. |
| 7. Resuspension & Assessment | Gently resuspend the cell pellet in fresh, pre-warmed complete culture medium. Perform a cell count and viability assessment (e.g., Trypan Blue exclusion). | Determines the success of the cryopreservation and thawing process. Low viability may indicate issues with freezing, thawing, or CPA toxicity [4]. |
The following workflow diagram illustrates the key decision points and procedures in this aseptic thawing process.
Post-Thaw Processing and Culture
After thawing and resuspension, plate the MSCs at an appropriate density. For complex 3D biofabricated constructs, the post-thaw recovery phase is critical. Research indicates that using cryoprotective biomaterials like hyaluronic acid (HA) or alginate in the scaffold can help maintain structural integrity and support post-thaw functionality by attenuating intracellular stress pathways [31]. Monitor the cultures closely for signs of contamination, such as rapid pH change or cloudiness in the media.
Advanced Strategies for Contamination Control
DMSO-Free Cryopreservation
A significant advancement in mitigating contamination risks associated with CPA toxicity is the move towards DMSO-free cryopreservation solutions. DMSO is cytotoxic and its presence in final cell products can trigger adverse reactions in patients [4] [81]. New fully defined, protein-free solutions are now available that eliminate DMSO, thereby reducing safety concerns and avoiding the potential compromise of cell function [81]. These kits often use a combination of penetrating and non-penetrating osmolytes to protect cells throughout the freeze-thaw cycle, resulting in post-thaw viability, attachment, proliferation, and differentiation potential comparable to traditional DMSO-containing media [81].
Environmental Monitoring and Risk Assessment
Proactive monitoring is essential for a robust contamination control strategy.
- Microbiological Risk Assessment: Employ tools like the Microbiological Risk Classification and Assessment (MiRCA) tool to systematically evaluate risks during the procurement and processing of cells and tissues. This tool helps identify critical control points, such as processing steps with a high percentage of post-processing microbiological findings [80].
- Environmental Monitoring: Regularly perform particle counts, aeroscopy (air sampling), and surface swabbing in critical areas like BSCs and cleanrooms to verify the aseptic environment [80]. Process cells in a Grade A cleanroom with a class B background as a standard for clinical-grade products [80].
The Scientist's Toolkit: Essential Research Reagents & Materials
The table below lists key materials and their functions for implementing the aseptic techniques described in this note.
Table 3: Essential Research Reagents and Materials for Aseptic Cryopreservation Work
| Item | Function & Application | Aseptic Consideration |
|---|---|---|
| DMSO-Free Cryopreservation Kit(e.g., CellShield MSC) | A fully defined, protein-free solution for freezing MSCs without the cytotoxicity and safety concerns of DMSO [81]. | Eliminates need to handle and dispose of cytotoxic DMSO; improves safety profile for eventual clinical use. |
| Pre-Sterilized Single-Use Consumables(e.g., pipettes, tubes, filters) | Acts as a primary barrier to contaminants; eliminates variability and potential failure of in-house sterilization [78]. | Critical for ensuring initial sterility; reduces workload and validation requirements for glassware cleaning. |
| Laminar Flow BSC / Biosafety Cabinet | Provides a HEPA-filtered, ISO 5 (Class 100) sterile environment for open manipulations of cells and media [78]. | The cornerstone of aseptic technique for all thawing and cell culture procedures. |
| Controlled-Rate Freezer (CRF) | Allows precise control of cooling rates during freezing, which is a critical process parameter for cell quality [74]. | Prevents ice crystal formation that can physically damage cells and constructs, compromising sterility barriers. |
| Validated Controlled-Thawing Device | Provides a GMP-compliant method for rapid, uniform thawing of cryovials, replacing contaminating water baths [74]. | Significantly reduces the risk of microbial contamination introduced during the thawing step. |
| Liquid Nitrogen Storage System(Vapor Phase) | Provides long-term storage at ≤ -150°C for preserved samples while minimizing cross-contamination risks [80]. | Storing in the vapor phase, rather than submerged in liquid, reduces the risk of liquid nitrogen penetrating vial seals. |
| Microbiological Risk Assessment Tool(e.g., MiRCA tool) | A systematic tool for identifying and quantifying microbiological risks in the cell processing workflow [80]. | Enables data-driven decisions to mitigate contamination risks at their source. |
For mesenchymal stem cell (MSC)-based tissue-engineered structures to achieve clinical success, cryopreservation must preserve not only cell viability but, crucially, their functional potency. Post-thaw functional potency—specifically the capacity for multilineage differentiation and immunomodulation—is the true benchmark for therapeutic efficacy in regenerative medicine [83] [27]. These parameters are delicate and can be compromised by cryoinjury, osmotic stress, and cryoprotectant agent (CPA) toxicity during the freeze-thaw cycle [27] [9]. This Application Note provides detailed protocols and analytical frameworks to ensure that cryopreserved MSC-based constructs retain their critical biological functions, thereby supporting the advancement of robust and reliable tissue-engineered therapies.
Critical Pre-Thaw Parameters and Cryopreservation Methods
The foundation of post-thaw functional potency is established during pre-thaw processing and the selection of an appropriate cryopreservation protocol. Key parameters including cell source, passage number, and culture history significantly influence the cryogenic resilience of MSCs [83] [1]. Furthermore, the choice between slow freezing and vitrification presents a trade-off between practicality and potential ice crystal damage [27].
Table 1: Comparison of Primary Cryopreservation Methods for MSCs
| Method | Mechanism | Cooling Rate | Key Advantage | Key Limitation | Reported Post-Thaw Viability |
|---|---|---|---|---|---|
| Slow Freezing | Gradual cellular dehydration; minimizes intracellular ice [27]. | ~ -1°C/min to -3°C/min [27]. | Operational simplicity, suitable for large volumes [27]. | Risk of solute damage and osmotic shock [27]. | 70-80% [27] |
| Vitrification | Ultra-rapid cooling to a glassy state; no ice crystal formation [27]. | Extremely high (> -1000°C/min) [27]. | Avoids mechanical damage from ice crystals. | Requires high [27], potentially toxic CPA concentrations; technique-sensitive. | Highly variable, depends on protocol optimization. |
Detailed Protocol: Slow Freezing of MSC Suspensions
The slow freezing method remains the most widely adopted technique for clinical-grade MSCs due to its scalability and relative ease of use [27]. The following protocol is optimized for preserving MSC function.
- Step 1: Preparation and Harvesting
- Use MSCs at early passages (P3-P5) that have been confirmed for standard surface marker expression (CD73+, CD90+, CD105+, CD14-, CD19-, CD34-, CD45-, HLA-DR-) and baseline differentiation potential [1].
- At ~80% confluence, harvest cells using a standard trypsinization protocol. Inactivate trypsin with complete culture medium.
- Step 2: CPA Addition and Equilibration
- Centrifuge the cell suspension and resuscle the pellet in a pre-chilled (4°C) cryopreservation solution. A common clinical-ready formulation is Plasmalyte A supplemented with 5% Human Albumin and 10% DMSO (PHD10) [84]. Alternatively, commercial solutions like CryoStor CS10 (10% DMSO) or CS5 (5% DMSO) can be used.
- A critical parameter is the final cell concentration. Data suggests MSCs can be cryopreserved at concentrations up to 9 million cells/mL without significant loss in viability or recovery, which allows for subsequent dilution of DMSO prior to infusion [84].
- Gently mix the cell-CPA suspension and aliquot into cryogenic vials.
- Equilibrate the vials on ice or at 4°C for 15-30 minutes to allow for CPA permeation.
- Step 3: Controlled-Rate Freezing
- Place the vials in a programmed controlled-rate freezer. A standard cooling ramp is -1°C per minute from 4°C to -80°C [27].
- If a controlled-rate freezer is unavailable, use a "Mr. Frosty"-type passive cooling device filled with isopropanol, which provides an approximate cooling rate of -1°C/min when placed at -80°C.
- Step 4: Long-Term Storage
- After completing the freezing ramp, immediately transfer the vials to the vapor phase of liquid nitrogen (-150°C to -196°C) for long-term storage [27].
The Scientist's Toolkit: Essential Reagents for Cryopreservation
Table 2: Key Research Reagent Solutions for MSC Cryopreservation
| Reagent Solution | Composition | Primary Function | Clinical Relevance |
|---|---|---|---|
| PHD10 | Plasmalyte-A, 5% Human Albumin, 10% DMSO [84]. | A clinically-ready formulation; DMSO acts as a penetrating CPA, albumin provides membrane stability and osmotic support. | High; components are suitable for human administration. |
| CryoStor CS10/CS5 | Proprietary, serum-free solutions with 10% or 5% DMSO and other undisclosed components [84]. | Optimized, standardized commercial formulations designed to minimize cryo-injury and improve post-thaw function. | High; GMP-manufactured, widely used in clinical trials. |
| NutriFreez D10 | Proprietary solution containing 10% DMSO [84]. | Ready-to-use commercial cryopreservation medium. | High; designed for clinical cell therapy applications. |
| DMSO (Laboratory Grade) | 100% Dimethyl Sulfoxide, diluted in culture medium or saline to 5-10%. | Penetrating CPA; reduces ice crystal formation by hydrogen bonding with water [9] [84]. | Requires washing or dilution prior to infusion due to potential patient toxicity [27] [84]. |
| Trehalose | Disaccharide sugar (often used at 0.2-0.5M) [9]. | Non-penetrating CPA; stabilizes cell membranes and proteins osmotically during freeze-thaw [9]. | Promising for DMSO-reduction strategies; considered biologically inert. |
Diagram 1: MSC Cryopreservation Workflow
Post-Thaw Viability and Potency Assessment
A comprehensive post-thaw assessment strategy must move beyond simple viability to confirm the retention of critical therapeutic functions.
Thawing and Immediate Analysis
- Thawing Protocol: Thaw cryopreserved vials rapidly by gentle agitation in a 37°C water bath until only a small ice crystal remains [27]. To mitigate contamination risk from a water bath, consider using dry heating equipment [27].
- CPA Removal and Dilution: Immediately after thawing, dilute the cell suspension drop-wise with a pre-warmed solution such as Plasmalyte A/5% Human Albumin to reduce the concentration of DMSO. A 1:2 dilution is effective when thawing cells cryopreserved at high concentration (e.g., 9 million cells/mL) [84]. Centrifuge gently to remove the CPA-containing supernatant.
- Viability and Recovery Quantification:
- Viability: Assess immediately post-thaw (T=0) and over a 6-hour period at room temperature to simulate clinical hold times. Use Trypan Blue exclusion for a rapid estimate and Annexin V/Propidium Iodide (PI) staining with flow cytometry to distinguish between live, early apoptotic, and dead cells [84].
- Recovery: Calculate viable cell recovery with the formula:
(Total Live Cells Counted / Number of Cells Originally Cryopreserved) * 100%[84]. Studies show that MSCs cryopreserved in 10% DMSO-based solutions (PHD10, NutriFreez) maintain comparable viabilities and recoveries for up to 6 hours post-thaw [84].
Protocol: Assessing Multilineage Differentiation Capacity
The gold standard for confirming retained differentiation potential involves directing post-thaw, culture-recovered MSCs toward osteogenic, adipogenic, and chondrogenic lineages [83] [1].
- Post-Thaw Culture: Plate the thawed and washed MSCs at a standard density (e.g., 5,000 cells/cm²) and culture for 3-6 days to allow for recovery. Proceed with differentiation once cells reach ~80% confluence.
- Osteogenic Differentiation:
- Induction Medium: Use Dulbecco's Modified Eagle Medium (DMEM) high glucose, supplemented with 10% FBS, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 50 µM ascorbate-2-phosphate.
- Protocol: Culture cells in induction medium for 21 days, changing the medium every 3-4 days.
- Analysis: Fix cells and stain with 2% Alizarin Red S (pH 4.1-4.3) to detect calcium deposits and mineralized matrix [1].
- Adipogenic Differentiation:
- Induction Medium: Use DMEM high glucose with 10% FBS, 1 µM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 10 µg/mL insulin, and 200 µM indomethacin.
- Protocol: Culture cells in induction medium for 14-21 days, with medium changes every 3-4 days.
- Analysis: Fix cells and stain with Oil Red O in 60% isopropanol to visualize intracellular lipid droplets [1].
- Chondrogenic Differentiation:
- Induction Medium: Use a high-density pellet culture system. Centrifuge 2.5x10⁵ cells to form a pellet. Culture pellets in DMEM high glucose supplemented with 1% ITS+ Premix, 100 nM dexamethasone, 50 µM ascorbate-2-phosphate, 40 µg/mL L-proline, and 10 ng/mL TGF-β3.
- Protocol: Culture pellets for 21-28 days, changing the medium every 2-3 days.
- Analysis: Fix pellets, paraffin-embed, section, and stain with Alcian Blue or Toluidine Blue to detect sulfated proteoglycans in the extracellular matrix [1].
Protocol: Evaluating Immunomodulatory Function
The therapeutic effect of MSCs is largely attributed to their potent immunomodulatory capabilities, which must be preserved post-thaw [85] [19]. Two key functional assays are detailed below.
- T Cell Proliferation Suppression Assay:
- Isolate PBMCs: Isolate peripheral blood mononuclear cells (PBMCs) from healthy donor buffy coats using Ficoll density gradient centrifugation.
- Label PBMCs: Label PBMCs with a cell proliferation dye such as Carboxyfluorescein succinimidyl ester (CFSE).
- Activate T Cells: Stimulate CFSE-labeled PBMCs (responder cells) with a mitogen like phytohemagglutinin (PHA) or with anti-CD3/CD28 beads to activate T cells.
- Co-culture Setup: Plate recovered, post-thaw MSCs (stimulator cells) and co-culture them with the activated PBMCs at varying MSC:PBMC ratios (e.g., 1:5, 1:10) for 4-5 days.
- Flow Cytometry Analysis: Harvest PBMCs and analyze CFSE dilution by flow cytometry to measure T cell proliferation. Compare the proliferation in co-culture with MSCs to the proliferation of PBMCs alone. Effective immunomodulation is indicated by a significant reduction in the percentage of proliferating (CFSE-low) T cells [19] [84].
- Macrophage Polarization Assay:
- Differentiate Macrophages: Differentiate isolated human monocytes into M0 macrophages using 50-100 ng/mL Macrophage Colony-Stimulating Factor (M-CSF) for 5-7 days.
- Polarize to M1 Phenotype: Polarize M0 macrophages toward a pro-inflammatory M1 phenotype using 100 ng/mL Lipopolysaccharide (LPS) and 20 ng/mL Interferon-gamma (IFN-γ) for 24-48 hours.
- Co-culture with MSCs: Establish a transwell co-culture system with M1 macrophages in the insert and post-thaw MSCs in the bottom well. Co-culture for 48-72 hours.
- Analysis: Analyze macrophage phenotype by flow cytometry. A shift toward an anti-inflammatory M2 phenotype is indicated by increased expression of markers like CD206 and CD163. Alternatively, measure the concentration of secreted cytokines in the supernatant; a decrease in TNF-α and an increase in IL-10 confirm functional immunomodulation [85] [19].
Diagram 2: MSC Immunomodulation Pathways
Quantitative Data and Functional Correlations
Rigorous quantification of post-thaw function is essential for protocol validation and quality control. The following table summarizes key findings from recent investigations.
Table 3: Impact of Cryopreservation Formulation on Post-Thaw MSC Quality Parameters
| Cryopreservation Solution | Reported Viability (0-6h Post-Thaw) | Cell Recovery Trend | Proliferative Capacity (Post-Recovery) | Immunomodulatory Potency (T cell suppression) | Key Findings |
|---|---|---|---|---|---|
| PHD10 (5% HA, 10% DMSO) | Comparable to other 10% DMSO solutions; stable over 6h [84]. | Maintained with optimal dilution [84]. | Similar to fresh cells/NutriFreez after 6-day culture [84]. | Preserved; comparable to NutriFreez [84]. | A robust, clinically-ready in-house formulation. |
| NutriFreez D10 (10% DMSO) | Comparable to PHD10; stable over 6h [84]. | Maintained with optimal dilution [84]. | Similar to PHD10 after 6-day culture [84]. | Preserved; comparable to PHD10 [84]. | Effective commercial option. |
| CryoStor CS10 (10% DMSO) | Comparable to other 10% DMSO solutions [84]. | Maintained with optimal dilution [84]. | 10-fold decrease vs. PHD10/NutriFreez at 3 & 6 M/mL [84]. | Not specified in search results, but low proliferation may impact long-term function. | Potential concern for applications requiring in vivo expansion. |
| CryoStor CS5 (5% DMSO) | Decreasing trend in viability and recovery noted [84]. | Lower trend vs. 10% DMSO formulations [84]. | 10-fold decrease vs. PHD10/NutriFreez at 3 & 6 M/mL [84]. | Not specified in search results. | Lower DMSO does not automatically equate to better functional preservation. |
Ensuring the functional potency of MSCs after cryopreservation is a multifaceted challenge that requires a meticulously optimized and standardized protocol. The data and methods presented herein demonstrate that the choice of cryopreservation solution, cell concentration, and post-thaw handling are critical determinants of success. While viability is a necessary first check, it is an insufficient metric alone. A thorough quality control regimen must include robust assays for differentiation capacity and immunomodulatory function—the true indicators of therapeutic potential. By adopting the detailed application notes and protocols outlined in this document, researchers and drug development professionals can significantly enhance the reliability and efficacy of MSC-based tissue-engineered structures, accelerating their translation from the bench to the clinic.
The transition from fetal bovine serum (FBS)-based to serum-free and xeno-free cryopreservation media represents a critical advancement in the clinical translation of Mesenchymal Stromal Cell (MSC)-based therapies. Traditional cryopreservation protocols relying on FBS raise significant concerns regarding batch-to-batch variability, risk of xenogeneic pathogen transmission, and undesirable immune responses in recipients [86] [87]. For MSC-based tissue-engineered structures, such as scaffold-free constructs, these risks are compounded, making the development of chemically defined, serum-free alternatives essential for manufacturing safe, consistent, and effective off-the-shelf regenerative products [88] [89].
This document provides detailed application notes and protocols for implementing advanced serum-free and xeno-free cryopreservation strategies, framed within the context of a broader thesis on preserving MSC-based tissue-engineered structures. It is designed to equip researchers and drug development professionals with the practical tools needed to navigate this complex landscape, ensuring the functional integrity of their cellular products post-thaw.
Performance Comparison of Serum-Free Cryopreservation Media
Selecting an appropriate serum-free medium requires careful consideration of its composition and performance. The following table summarizes key functional data from a comprehensive study evaluating commercially available, animal-protein-free freezing media for peripheral blood mononuclear cells (PBMCs), providing a valuable reference for MSC cryopreservation [86].
Table 1: Viability and Functionality of Cells Cryopreserved in Serum-Free Media Over 24 Months [86]
| Freezing Medium | DMSO Concentration | Viability (Over 24 Months) | T-cell Functionality | B-cell Functionality | Recommended for Long-Term Storage? |
|---|---|---|---|---|---|
| CryoStor CS10 | 10% | High (>80%, comparable to FBS control) | Preserved | Preserved | Yes |
| NutriFreez D10 | 10% | High (>80%, comparable to FBS control) | Preserved | Preserved | Yes |
| Bambanker D10 | 10% | High | Tendency for divergence from FBS control | Preserved | With Caution |
| CryoStor CS7.5 | 7.5% | Promising at early time points | N/A | N/A | No (Eliminated from study) |
| Media with <7.5% DMSO | 2%-5% | Significant loss | Not maintained | Not maintained | No |
The data conclusively shows that media containing 10% DMSO, specifically CryoStor CS10 and NutriFreez D10, consistently maintain high cell viability and functionality equivalent to the traditional FBS-based reference medium for up to 24 months. Media with reduced DMSO concentrations (below 7.5%) were found to be inadequate for long-term preservation, resulting in significant viability loss [86]. This underscores the current necessity of DMSO as an effective cryoprotectant, even in advanced serum-free formulations.
Protocols for Serum-Free Cryopreservation of MSCs and Constructs
Cryopreservation of Monolayer MSCs
This protocol is adapted for the preservation of research-grade MSC monolayers using serum-free media [86] [4].
Materials:
- Cells: MSC monolayers at 70-80% confluence.
- Freezing Media: Pre-warmed, commercially available serum-free freezing medium (e.g., CryoStor CS10, NutriFreez D10).
- Reagents: Phosphate-Buffered Saline (PBS), TrypLE Select or other xeno-free dissociation enzyme.
- Supplies: Cryogenic vials, controlled-rate freezing container (e.g., CoolCell), isopropanol.
Procedure:
- Harvesting: Aspirate the culture medium and wash the monolayer gently with PBS. Detach the cells using a xeno-free dissociation enzyme according to the manufacturer's instructions. Neutralize the enzyme with an appropriate serum-free medium.
- Centrifugation: Centrifuge the cell suspension at 300 × g for 5 minutes. Aspirate the supernatant completely.
- Resuspension: Resuspend the cell pellet in the pre-warmed serum-free freezing medium at a density of 1-5 × 10^6 cells/mL. Gently mix to ensure a homogeneous suspension.
- Aliquoting: Dispense 1 mL of the cell suspension into each cryogenic vial. Place the vials immediately on wet ice or in a 4°C refrigerator.
- Freezing: Transfer the vials into a controlled-rate freezing container pre-cooled to 4°C. Place the container immediately in a -80°C freezer for a minimum of 24 hours. The controlled-rate freezer ensures a consistent cooling rate of approximately -1°C/min, which is critical for high viability.
- Long-Term Storage: After 24 hours, promptly transfer the cryovials to a liquid nitrogen storage tank for long-term preservation.
Cryopreservation of Scaffold-Free Tissue-Engineered Constructs
This protocol outlines the methodology for cryopreserving three-dimensional scaffold-free constructs, such as the "gMSC1" sheet-like TEC, as described in recent literature [88].
Materials:
- Constructs: Scaffold-free MSC sheets (e.g., gMSC1) cultured in serum-free media.
- Freezing Media: Serum-free cryopreservation medium (e.g., CryoStor CS10).
- Carrier Solution: Lactated Ringer's solution (e.g., Solulact).
- Supplies: Cryovials, controlled-rate freezing apparatus.
Procedure:
- Harvesting: Carefully detach the scaffold-free construct from the culture dish using a sterile pipette tip or spatula.
- Equilibration: Transfer the individual construct to a cryovial containing a mixture of Lactated Ringer's solution and the serum-free freezing medium. The study on gMSC1 demonstrated that this step is feasible and does not compromise the construct's post-thaw properties or in vivo efficacy [88].
- Freezing: Place the cryovials in a controlled-rate freezer, following a slow freezing protocol. While the specific cooling curve for gMSC1 was not detailed, standard slow freezing protocols (approximately -1°C/min) are applicable.
- Storage: Store the vials in the vapor phase of liquid nitrogen.
- Thawing and Washing: Rapidly thaw the constructs in a 37°C water bath. Gently wash the constructs with a serum-free culture medium to remove the cryoprotectant before implantation or further analysis. Studies confirm that frozen gMSC1 (Fro-gMSC1) retains its cartilage repair capacity, filling defects with cartilage-like tissue that integrates well with adjacent host tissue [88].
The diagram below illustrates the core workflow and decision-making process for the cryopreservation protocols described above.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of serum-free cryopreservation relies on a suite of specialized reagents and equipment. The following table details key solutions and their functions in the process.
Table 2: Essential Research Reagent Solutions for Serum-Free Cryopreservation
| Item | Function & Rationale | Example Products / Notes |
|---|---|---|
| Serum-Free Freezing Medium | Provides a defined, xeno-free matrix with cryoprotectants and nutrients to protect cells during freeze-thaw cycles, eliminating FBS-associated risks. | CryoStor CS10, NutriFreez D10 [86]. |
| Xeno-Free Dissociation Enzyme | Gently dissociates adherent MSCs without using animal-derived trypsin, maintaining a completely xeno-free workflow. | TrypLE Select [88]. |
| Controlled-Rate Freezing Device | Ensures a consistent, optimal cooling rate (typically ~-1°C/min), which is critical for high cell survival and reproducible results. | CoolCell, Mr. Frosty, or programmable freezer [29] [4]. |
| Cryogenic Storage Vials | Secure, leak-proof containers designed to withstand extreme temperatures of liquid nitrogen storage. | Internally-threaded vials are recommended to prevent contamination. |
| Defined Culture Media | For post-thaw culture and expansion; serum-free media (e.g., STK2, MSC-Brew GMP) are essential to maintain the xeno-free status of the cells. | STK2 [88], MSC-Brew GMP [89], MesenCult-ACF [87]. |
Critical Considerations for Media Selection and Functional Validation
Navigating Media Selection and Performance
While serum-free media offer significant advantages, their performance can vary. Research indicates that not all serum-free media are created equal; some commercially available "serum-free" media may still contain significant levels of human-derived components like platelet lysate, which reintroduces variability [90]. Furthermore, a medium optimized for rapid MSC proliferation does not necessarily support the cells' therapeutic potency, such as chondrogenic capacity for cartilage repair [87]. Therefore, the choice of medium must be validated against the specific functional application of the final MSC product.
Post-Thaw Functional Validation Assays
Simply measuring viability post-thaw is insufficient. A comprehensive quality control assessment must include functional assays to ensure the preserved cells retain their therapeutic potential.
- In Vitro Differentiation: Confirm retained capacity for trilineage differentiation (osteogenic, adipogenic, chondrogenic) as per ISCT guidelines [3] [4].
- Immunomodulatory Potency: Assess the ability of thawed MSCs to suppress the activation of T-lymphocytes in co-culture assays [89].
- In Vivo Efficacy: For tissue-engineered constructs, the ultimate validation is the demonstration of functional efficacy in an appropriate animal model, such as the successful repair of cartilage defects in a rat model by frozen gMSC1 constructs [88] [29].
- Phenotype and Senescence: Analyze surface marker expression (CD73, CD90, CD105) and assess senescence-associated markers (e.g., β-galactosidase activity) to ensure the cells have not undergone significant phenotypic alteration or aging during the process [87].
The following diagram outlines the key decision-making pathway and critical checks for selecting and validating a serum-free cryopreservation medium.
Assessing Efficacy, Safety, and Clinical Potential of Cryopreserved Products
The transition of Mesenchymal Stromal Cell (MSC)-based therapies from research to clinical application hinges on the establishment of robust, standardized quality control metrics. For tissue-engineered structures, where cells are integrated with biomaterial scaffolds, the post-thaw recovery of MSCs is particularly critical. The cryopreservation process must preserve not only basic cell viability but also key functional characteristics that enable MSCs to perform their therapeutic roles. This application note details four essential quality metrics—viability, apoptosis, attachment efficiency, and population doubling—providing standardized protocols and benchmark data to ensure the consistent quality of MSC-based products in regenerative medicine and drug development.
Essential Quality Metrics: Protocols and Benchmark Data
Regular monitoring of these metrics post-thaw is crucial for validating cryopreservation protocols and ensuring that MSCs maintain their therapeutic potential for tissue engineering applications.
Viability
Purpose: Viability measurement determines the proportion of live cells in a population immediately after thawing, serving as the most fundamental indicator of cryopreservation success.
Experimental Protocol (Trypan Blue Exclusion):
- Thawing: Rapidly thaw cryopreserved MSCs in a 37°C water bath until only a small ice crystal remains.
- Dilution: Immediately dilute the cell suspension 1:10 with pre-warmed complete culture medium to reduce cryoprotectant toxicity.
- Centrifugation: Centrifuge the cells at 300 × g for 5 minutes and carefully aspirate the supernatant containing the cryopreservation medium [55].
- Staining: Resuspend the cell pellet in an appropriate volume of PBS. Mix 10 µL of cell suspension with 10 µL of 0.4% Trypan Blue solution and incubate for 1-3 minutes.
- Counting: Load the mixture onto a hemocytometer. Count both unstained (viable) and blue-stained (non-viable) cells. Calculate viability percentage: (Number of viable cells / Total number of cells) × 100.
Industry Standards: Research-grade applications typically require post-thaw viability >70%, while clinical applications under Good Manufacturing Practice (GMP) standards demand >95% viability for product release [91] [92].
Apoptosis
Purpose: Apoptosis assessment detects early-stage programmed cell death, which can be triggered by cryopreservation-induced stresses and may not be immediately evident through viability assays alone.
Experimental Protocol (Flow Cytometry with Annexin V/PI):
- Sample Preparation: At 24 hours post-thaw, harvest approximately 1×10^5 MSCs by gentle trypsinization.
- Washing: Wash cells twice with cold PBS and resuspend in 1X Annexin V binding buffer.
- Staining: Transfer 100 µL of cell suspension to a flow cytometry tube. Add 5 µL of fluorochrome-conjugated Annexin V and 5 µL of Propidium Iodide (PI) staining solution.
- Incubation: Incubate for 15 minutes at room temperature in the dark.
- Analysis: Add 400 µL of binding buffer and analyze within one hour using flow cytometry. Use the following gating: Annexin V-/PI- (viable), Annexin V+/PI- (early apoptotic), Annexin V+/PI+ (late apoptotic/necrotic).
Interpretation: A successful cryopreservation protocol should result in >85% viable cells (Annexin V-/PI-) and <10% early apoptotic cells (Annexin V+/PI-) at 24 hours post-thaw.
Attachment Efficiency
Purpose: This metric evaluates the functional capacity of cryopreserved MSCs to adhere to culture surfaces or biomaterial scaffolds—a critical prerequisite for in vitro expansion and in vivo engraftment in tissue-engineered constructs.
Experimental Protocol:
- Seeding: After thawing and determining viability, seed MSCs at a low density (5-10 cells/mm²) in standard culture vessels or onto representative scaffold materials.
- Incubation: Allow cells to attach for 24 hours under standard culture conditions (37°C, 5% CO₂).
- Washing: Gently wash the surface with PBS to remove non-adherent cells.
- Fixation and Staining: Fix attached cells with 4% paraformaldehyde for 15 minutes and stain with crystal violet or calcein AM.
- Quantification: Count attached cells manually or using automated imaging systems. Calculate attachment efficiency: (Number of attached cells / Number of cells seeded) × 100.
Benchmark Data: High-quality cryopreserved MSCs should demonstrate ≥80% attachment efficiency compared to non-frozen controls when assessed 24 hours post-thaw.
Population Doubling
Purpose: Population doubling assessment evaluates the proliferative capacity of MSCs after cryopreservation, ensuring they retain their growth potential for expansion and tissue formation.
Experimental Protocol (Population Doubling Time):
- Seeding: After determining attachment efficiency at 24 hours, harvest and count attached cells. Seed a known number of cells (e.g., 5 × 10³ cells/cm²) in fresh culture vessels [91].
- Harvesting: Culture cells until they reach 80-90% confluency, then harvest and count again.
- Calculation: Calculate population doubling time using the formula: Doubling Time = (Duration of culture × Log(2)) / (Log(Final cell count) - Log(Initial cell count)) where duration is in hours [91].
Quality Benchmark: Cryopreserved MSCs should maintain a consistent doubling time across passages that is comparable to non-frozen controls, typically ranging between 24-72 hours depending on the MSC source and donor characteristics.
Table 1: Quality Metric Benchmarks for Cryopreserved MSCs
| Quality Metric | Assessment Method | Time Point | Research Grade Standard | Clinical Grade Standard | Key Considerations |
|---|---|---|---|---|---|
| Viability | Trypan Blue Exclusion | 0-2 hours post-thaw | >70% | >95% [91] | Use validated automated cell counters for consistency |
| Apoptosis | Annexin V/PI Flow Cytometry | 24 hours post-thaw | <15% early apoptotic cells | <10% early apoptotic cells | Distinguishes early apoptosis from necrosis |
| Attachment Efficiency | Crystal Violet Staining | 24 hours post-thaw | ≥70% | ≥80% | Critical for scaffold-based tissue engineering |
| Population Doubling Time | Sequential Cell Counting | 3-5 days post-thaw | Consistent with pre-freeze values | Consistent with pre-freeze values | Indicator of long-term functional recovery |
Quality Assessment Workflow
The following diagram illustrates the recommended temporal workflow for assessing the four key quality metrics of cryopreserved MSCs, from immediate post-thaw analysis to evaluation of long-term functionality.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for MSC Quality Assessment
| Reagent/Cell Culture Material | Function | Example Products | GMP-Grade Requirement |
|---|---|---|---|
| Defined Cryopreservation Medium | Protects cells during freezing/thawing; reduces ice crystal formation | CryoStor CS10, MesenCult-ACF Freezing Medium [55] | Required for clinical applications |
| Animal Component-Free Culture Media | Supports MSC growth without animal-derived components; enhances clinical safety | MSC-Brew GMP Medium, MesenCult-ACF Plus Medium [91] | Required for clinical applications |
| Annexin V/Propidium Iodide Kit | Distinguishes viable, early apoptotic, and late apoptotic/necrotic cell populations | BD Pharmingen Annexin V Apoptosis Detection Kits | Recommended for clinical applications |
| Collagenase Solution | Tissue digestion for primary MSC isolation from source tissues | GMP-grade Collagenase | Required for clinical applications |
| Flow Cytometry Antibody Panels | Confirmation of MSC identity (CD73+, CD90+, CD105+, CD34-, CD45-, HLA-DR-) | BD Stemflow Human MSC Analysis Kit [91] | Required for clinical applications |
| Cell Culture Vessels/Scaffolds | Assessment of attachment efficiency on culture surfaces or biomaterials | Tissue culture-treated plastics, engineered scaffolds | Dependent on application |
The systematic assessment of viability, apoptosis, attachment efficiency, and population doubling provides a comprehensive framework for evaluating the success of MSC cryopreservation protocols. For tissue-engineered structures, where MSCs must not only survive freezing but also maintain their functional capacity to integrate with biomaterials and participate in tissue regeneration, these metrics are indispensable. By implementing the standardized protocols and benchmark values outlined in this application note, researchers and drug development professionals can significantly enhance the reproducibility, efficacy, and safety of MSC-based therapies, ultimately accelerating their translation from the laboratory to clinical applications.
Within the broader scope of cryopreservation research for mesenchymal stromal cell (MSC)-based tissue-engineered structures, functional potency assays remain a critical benchmark for evaluating post-thaw cellular integrity. Cryopreservation is an indispensable step for the off-the-shelf availability of advanced therapy medicinal products (ATMPs), yet the freezing and thawing processes can induce cellular stress, potentially compromising therapeutic efficacy [93]. While phenotypic marker analysis provides basic quality data, trilineage differentiation potential—specifically, the capacity for osteogenic, adipogenic, and chondrogenic differentiation—serves as a fundamental functional correlate for stemness and regenerative capability [4] [94]. This document presents standardized application notes and detailed protocols for quantifying the differentiation potency of MSCs following cryopreservation, providing a critical toolset for ensuring the functional quality of tissue-engineered constructs in regenerative medicine.
The Impact of Cryopreservation on MSC Potency
The process of cryopreservation, while necessary for storage and distribution, imposes significant stress on MSCs. The formation of intra- and extracellular ice crystals, osmotic shock, and cryoprotectant agent (CPA) toxicity can deleteriously affect cell viability, recovery, and, most importantly, function [4] [93]. Table 1 summarizes key functional attributes that can be impacted by the freeze-thaw cycle.
Table 1: Functional Attributes of MSCs Affected by Cryopreservation
| Functional Attribute | Reported Impact of Cryopreservation | Relevant Citation |
|---|---|---|
| Immunomodulatory Capacity | Decreased responsiveness to IFN-γ, reduced IDO activity, and impaired T-cell suppression immediately post-thaw; often recovers after 24h acclimation. | [95] [96] |
| Angiogenic Secretome | Secretion of angiogenic cytokines (e.g., VEGF, HGF, IL-8) may be maintained in some cryopreservation formats. | [96] |
| Proliferation & Metabolism | Significant decrease in cell proliferation and metabolic activity immediately post-thaw. | [95] |
| Apoptosis | Significant increase in early and late apoptosis/necrosis in freshly thawed cells. | [95] |
| Trilineage Differentiation | Multipotent capacity is generally maintained, but quantitative potency can be diminished without a post-thaw recovery period. | [95] [94] |
A critical finding is that a post-thaw acclimation period can reverse many of these functional deficits. One study demonstrated that MSCs acclimated for 24 hours post-thaw (Thawed + Time, TT) showed significantly reduced apoptosis and upregulated expression of angiogenic and anti-inflammatory genes compared to those used immediately (Freshly Thawed, FT) [95]. This underscores the importance of incorporating a recovery phase into potency testing workflows to accurately assess the innate functional capacity of the cells, separate from transient cryopreservation-induced stress.
Quantitative Differentiation Potency Post-Thaw
The differentiation potential of MSCs is not uniform across all lineages and can be influenced by cell source, donor variability, and specific subpopulations. Furthermore, the cryopreservation process itself can have a differential impact on these capacities. Table 2 consolidates quantitative data from recent studies on the differentiation performance of various MSC populations, providing a benchmark for post-thaw potency.
Table 2: Quantitative Differentiation Performance of MSC Populations
| Cell Type / Population | Osteogenic Performance | Adipogenic Performance | Chondrogenic Performance | Citation |
|---|---|---|---|---|
| Sca-1+ ADSCs (ADSC-AM) | Supported (Specific quantitative data not provided) | Enhanced adipogenic differentiation | Supported (Specific quantitative data not provided) | [97] |
| Bone Marrow MSCs (Post-Thaw, Acclimated) | Positive Alizarin Red staining (Calcium deposits) | Not tested in cited study | Positive Alcian Blue staining (Proteoglycans) | [95] |
| CD146+ MFAT Cells | Significantly higher calcium deposition vs. CD34+, CD271+, and unsorted cells. | Not tested in cited study | Produced proteoglycans (Safranin-O+) | [98] |
| CD271+ MFAT Cells | Positive Alizarin Red staining (Calcium deposits) | Not tested in cited study | Greatest proteoglycan formation (Safranin-O+) vs. CD34+ and CD146+ cells. | [98] |
| ADSCs (Rosiglitazone + T3 Induced) | Not primary focus of study | Optimal brown adipogenesis: 19-fold ↑ UCP1, 7.5-fold ↑ PPARγ, 3.8-fold ↑ PGC1α | Not primary focus of study | [99] |
These findings highlight that the functional potency of a cryopreserved MSC product is a multi-faceted metric. A comprehensive potency assay matrix should therefore evaluate all three lineages to fully characterize the cell product, especially when derived from tissues like microfragmented adipose tissue (MFAT) where inherent heterogeneity is significant [98].
Experimental Protocols for Trilineage Differentiation
The following protocols are standardized for evaluating the potency of cryopreserved MSCs after a 24-hour post-thaw recovery period to ensure accurate assessment of baseline functionality [95].
Protocol 1: Osteogenic Differentiation
Principle: To induce and quantify MSC differentiation into osteoblasts by evaluating calcium phosphate deposition, a key marker of bone matrix formation.
Materials:
- Induction Medium: Dulbecco's Modified Eagle Medium (DMEM) high glucose, 10% Fetal Bovine Serum (FBS), 1% Penicillin-Streptomycin (P/S), 100 nM Dexamethasone, 10 mM β-glycerophosphate, 50 µM Ascorbic Acid-2-phosphate.
- Staining Solution: 2% Alizarin Red S, pH 4.1-4.3.
- Positive Control: Commercially available osteogenic differentiation kits (e.g., StemPro Osteogenesis Differentiation Kit).
Procedure:
- Seeding: Thaw and acclimate MSCs for 24 hours. Seed cells at a density of 3.1 x 10^3 cells/cm² in standard growth medium and allow to adhere overnight.
- Induction: Replace growth medium with osteogenic induction medium. Maintain cultures for 21 days, refreshing the induction medium twice weekly.
- Analysis (Post 21 days):
- Fixation: Aspirate medium, wash with PBS, and fix cells with 4% neutral buffered formalin for 15 minutes.
- Staining: Incubate with Alizarin Red S solution for 20-30 minutes at room temperature with gentle agitation.
- Washing & Imaging: Wash extensively with distilled water to remove non-specific stain and capture images.
- Quantification: For quantitative analysis, elute bound dye with 10% (w/v) cetylpyridinium chloride for 1 hour and measure absorbance at 562 nm [95] [98].
Protocol 2: Adipogenic Differentiation
Principle: To induce and quantify MSC differentiation into adipocytes by visualizing intracellular lipid droplet accumulation.
Materials:
- Induction Medium: DMEM high glucose, 10% FBS, 1% P/S, 1 µM Dexamethasone, 0.5 mM 3-Isobutyl-1-methylxanthine (IBMX), 100 µM Indomethacin, 10 µg/mL Insulin.
- Maintenance Medium: DMEM high glucose, 10% FBS, 1% P/S, 10 µg/mL Insulin.
- Staining Solution: Oil Red O working solution (0.3% in 60% isopropanol).
- For Brown Adipogenesis: Supplement with 1000 nM Rosiglitazone and 0.2 nM Triiodothyronine (T3) [99].
Procedure:
- Seeding: Seed post-thaw, acclimated MSCs at a density of 3.1 x 10^3 cells/cm² and allow to reach 100% confluence.
- Cyclic Induction: Initiate differentiation by exposing cells to adipogenic induction medium for 3-4 days, followed by maintenance medium for 1-3 days. Repeat this cycle 3-4 times.
- Analysis (Post 14-21 days):
- Fixation: Wash with PBS and fix cells with 4% formalin for 15-30 minutes.
- Staining: Incubate with Oil Red O working solution for 30-60 minutes.
- Washing & Imaging: Wash with distilled water and image. Multilocular lipid droplets are indicative of brown adipocytes [99].
- Quantification: Elute Oil Red O with 100% isopropanol and measure absorbance at 500-520 nm.
Protocol 3: Chondrogenic Differentiation
Principle: To induce and quantify MSC condensation and differentiation into chondrocytes, producing a cartilage-specific extracellular matrix rich in sulfated proteoglycans.
Materials:
- Induction Medium: DMEM high glucose, 1% ITS+ Supplement, 1% P/S, 100 nM Dexamethasone, 50 µg/mL Ascorbic Acid-2-phosphate, 40 µg/mL L-Proline, 10 ng/mL TGF-β3.
- Staining Solutions: 1% Alcian Blue in 3% acetic acid (pH 2.5) or Safranin-O.
Procedure (Micromass Culture):
- Pellet Formation: Resuspend 2.5 x 10^5 post-thaw, acclimated MSCs in 15 mL conical tube with chondrogenic induction medium. Centrifuge at 500 x g for 5 minutes to form a pellet.
- Induction: Loosen tube caps for gas exchange and culture pellets for 21-28 days, refreshing the induction medium every 2-3 days.
- Analysis (Post 21-28 days):
- Fixation & Sectioning: Fix pellets in 4% formalin, embed in paraffin, and section.
- Staining: Deparaffinized sections are stained with Alcian Blue (for acidic proteoglycans) or Safranin-O (for sulfated proteoglycans).
- Imaging & Quantification: Image sections. Pellet size and staining intensity can be quantified using image analysis software. qPCR for biomarkers like ACAN and COL2A1 is recommended for molecular confirmation [95] [98].
Signaling Pathways in MSC Differentiation
The differentiation of MSCs down osteogenic, adipogenic, and chondrogenic lineages is governed by highly regulated and distinct signaling pathways. Mastering these molecular mechanisms is crucial for developing targeted differentiation protocols and accurately interpreting potency assay results. The following diagram synthesizes the core signaling cascades involved in each lineage commitment.
Experimental Workflow for Post-Thaw Potency Assessment
A rigorous and standardized workflow is essential for generating reliable and reproducible data on the functional potency of cryopreserved MSCs. The following diagram outlines the key stages, from cell recovery post-thaw to final data analysis, integrating the critical 24-hour acclimation period that allows cells to recover from cryopreservation stress.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues critical reagents and their functions for successfully executing the post-thaw trilineage differentiation protocols outlined in this document.
Table 3: Essential Reagents for Trilineage Differentiation Assays
| Reagent/Category | Specific Examples | Function in Differentiation | Protocol |
|---|---|---|---|
| Cryopreservation | Cryostor CS5, DMSO, FBS | Cryoprotectant; prevents ice crystal formation and maintains cell viability during freeze-thaw. | Pre-Assay |
| Lineage Induction | Dexamethasone, β-glycerophosphate, Ascorbic Acid | Promotes osteoblast maturation and mineralization. | Osteogenic |
| Lineage Induction | IBMX, Indomethacin, Insulin, Rosiglitazone, T3 | Induces lipid accumulation and adipocyte maturation; T3/rosiglitazone enhances brown adipogenesis. | Adipogenic |
| Lineage Induction | TGF-β3, ITS+ Supplement, L-Proline | Key inducer of chondrogenesis; supports cartilage-specific matrix synthesis. | Chondrogenic |
| Histological Stains | Alizarin Red S | Binds to calcium phosphate, visualizes mineralized matrix. | Osteogenic |
| Histological Stains | Oil Red O | Stains neutral lipids and triglycerides in lipid droplets. | Adipogenic |
| Histological Stains | Alcian Blue, Safranin-O | Stains sulfated and acidic proteoglycans in cartilage matrix. | Chondrogenic |
| Molecular Analysis | qPCR primers for RUNX2, PPARγ, UCP1, ACAN | Quantifies gene expression of lineage-specific markers for mechanistic insights. | All |
The derivation of mesenchymal stem cells (MSCs) from human pluripotent stem cells (hPSCs) represents a promising approach for obtaining standardized cellular materials for regenerative medicine. However, the intrinsic tumorigenic potential of undifferentiated hPSCs poses a significant safety concern for clinical translation. Residual undifferentiated hPSCs within differentiated MSC populations can form teratomas—benign tumors containing derivatives of all three germ layers—following transplantation [100] [101] [102]. This Application Note outlines a comprehensive risk assessment strategy and experimental protocols to manage teratoma formation potential in hPSC-derived MSC products, framed within the context of cryopreservation research for tissue-engineered structures. Implementing robust sensitive detection methodologies is essential for ensuring patient safety and regulatory compliance for advanced therapy medicinal products (ATMPs) [100].
The tumorigenicity risk profile of hPSC-derived products depends on multiple factors, including the differentiation status, proliferation capacity, residual undifferentiated cell burden, and post-processing handling such as cryopreservation [103]. This document provides researchers and drug development professionals with standardized protocols for quantifying this risk, emphasizing the integration of these assessments with biobanking workflows to maintain product consistency and safety from development through clinical application.
Risk Assessment Framework
Understanding the Hazard
Pluripotent stem cells, including both embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), are defined by their capacity for unlimited self-renewal and ability to differentiate into derivatives of all three germ layers. This same pluripotency underlies their potential to form teratomas when transplanted into immunocompromised animal models [103]. The transition to a clinical MSC product requires complete departure from this pluripotent state, yet residual undifferentiated cells may persist through the differentiation process. Current regulatory frameworks require rigorous safety assessments to mitigate this tumorigenicity risk, traditionally relying on in vivo animal models [100] [101].
Key Risk Factors
Multiple parameters influence the overall risk profile of hPSC-derived MSC products:
- Cell Source: hPSC lines exhibit varying propensities for teratoma formation based on their genetic background, reprogramming method (for iPSCs), and culture history [104] [103].
- Differentiation Protocol Efficiency: Well-optimized, reproducible differentiation protocols minimize residual pluripotent cells [104].
- Purification Strategies: Specific surface marker-based isolation (e.g., CD105+, CD73+, CD90+, CD45-) can enrich for target MSC populations while depleting undifferentiated cells [4] [102].
- Cryopreservation Impact: Freeze-thaw cycles may selectively affect different cell subpopulations, potentially altering the ratio of undifferentiated to differentiated cells post-thaw [4] [105] [9].
- In Vitro Expansion: Extensive passaging increases the risk of genetic and epigenetic instability, potentially affecting both transformation risk and differentiation stability [106].
Table 1: Comparative Sensitivity of Tumorigenicity Assessment Methods
| Method | Detection Limit | Time to Result | Key Advantages | Key Limitations |
|---|---|---|---|---|
| In Vivo Teratoma Assay | ~1x10⁴ cells [100] | 12-20 weeks | Gold standard, provides pathological context | Low throughput, high cost, animal use, questionable translational relevance |
| Highly Efficient Culture (HEC) Assay | ≤1x10⁻⁶ [100] | 3-4 weeks | High sensitivity, quantitative, detects functional pluripotency | Requires specialized culture expertise |
| Digital PCR (ddPCR) | ≤1x10⁻⁶ [100] [101] | 1-2 days | High sensitivity, quantitative, reproducible, amenable to standardization | Detects molecular markers but not functional pluripotency |
| Flow Cytometry | ~1x10⁻⁴ [100] | 1 day | Rapid, can be multiplexed | Lower sensitivity, marker expression may vary |
Experimental Protocols
Sample Preparation and Cryopreservation
Principle: Standardized preparation and cryopreservation of hPSC-derived MSCs ensures consistent starting material for tumorigenicity assessment while mimicking clinical manufacturing workflows.
Reagents and Materials:
- hPSC-derived MSC cell suspension
- Cryoprotective medium: Culture medium supplemented with 10% DMSO and 10-20% serum or defined alternatives [4] [9]
- Programmable freezing container or controlled-rate freezer
- Liquid nitrogen storage system
Procedure:
- Harvesting: Detach hPSC-derived MSCs at appropriate confluence using enzyme-free dissociation buffer or low-concentration trypsin-EDTA.
- Quantification: Perform accurate cell counting and viability assessment using trypan blue exclusion or automated cell counters.
- Formulation: Centrifuge cells and resuspend in cryoprotective medium at 1-5×10⁶ cells/mL.
- Aliquoting: Dispense 1.0-1.5 mL aliquots into cryogenic vials.
- Controlled-Rate Freezing: Use a programmed freeze cycle of -1°C/min to -40°C, then -10°C/min to -90°C, followed by rapid transfer to liquid nitrogen vapor phase (-135°C to -196°C) for long-term storage [4] [105].
- Thawing: Rapidly thaw cryopreserved vials in a 37°C water bath until only a small ice crystal remains, then immediately transfer to pre-warmed culture medium containing gradual dilutions of DMSO to minimize osmotic shock.
Quality Control: Post-thaw viability should exceed 80% by dye exclusion assays. Cells should maintain characteristic MSC surface marker expression (CD105, CD73, CD90) and absence of hematopoietic markers (CD45, CD34, CD14) [4].
Highly Efficient Culture (HEC) Assay for Pluripotent Cell Detection
Principle: This highly sensitive in vitro method exploits the ability of residual undifferentiated hPSCs to form colonies under conditions that selectively promote pluripotent cell growth while suppressing differentiated cell types [100].
Reagents and Materials:
- Test article: hPSC-derived MSC preparation (fresh or post-thaw)
- Positive control: Parental hPSC line
- HEC medium: mTeSR1 or equivalent defined pluripotent stem cell medium
- Irradiated mouse embryonic fibroblasts (MEFs) or equivalent feeder layer
- 6-well tissue culture plates
Procedure:
- Feeder Layer Preparation: Plate irradiated MEFs at 1.5-2.0×10⁴ cells/cm² in 6-well plates and allow to attach overnight.
- Test Article Plating: Plate hPSC-derived MSCs in a dilution series from 1×10⁵ to 1×10⁷ cells per well in HEC medium.
- Positive Control Plating: Plate defined numbers of parental hPSCs (10-1000 cells) to establish standard curve and assay sensitivity.
- Culture Maintenance: Culture for 3-4 weeks with medium changes every other day, monitoring for colony formation.
- Colony Identification and Quantification: Score wells for the presence of pluripotent stem cell colonies characterized by defined borders, high nucleus-to-cytoplasm ratio, and prominent nucleoli.
- Validation: Fix and stain a subset of colonies for pluripotency markers (OCT4, NANOG, SSEA-4) to confirm identity.
Calculation: The frequency of residual undifferentiated cells is calculated using Poisson statistics: Frequency = -ln(P₀)/N, where P₀ is the proportion of negative wells and N is the number of cells plated per well [100].
Digital PCR (ddPCR) for Pluripotency Marker Detection
Principle: Droplet digital PCR provides absolute quantification of rare targets by partitioning samples into thousands of nanoliter-sized droplets and analyzing amplification in each droplet individually, enabling detection of pluripotency-associated transcripts at frequencies below 1 in 1 million cells [100] [101].
Reagents and Materials:
- Test article: hPSC-derived MSC RNA or cDNA
- Positive control: Parental hPSC RNA
- ddPCR Supermix for Probes
- Pluripotency marker assay: Pre-designed or custom primer/probe sets for OCT4 (POU5F1), NANOG, or other validated markers
- Housekeeping gene assay: GAPDH, HPRT1, or similar
- Droplet generator and reader
Procedure:
- RNA Isolation: Extract high-quality total RNA from test and control samples using silica-membrane columns with DNase treatment.
- Reverse Transcription: Convert equal amounts of RNA (100-500 ng) to cDNA using reverse transcriptase with random hexamers.
- Reaction Setup: Prepare 20µL reactions containing ddPCR Supermix, target assay (FAM-labeled), and reference assay (HEX-labeled).
- Droplet Generation: Generate 10,000-20,000 droplets per sample using the droplet generator.
- PCR Amplification: Run thermal cycling with optimized annealing/extension temperatures for target assays.
- Droplet Reading: Quantify fluorescence in each droplet using the droplet reader.
- Data Analysis: Use companion software to determine the absolute concentration (copies/µL) of target and reference genes in the original reaction.
Interpretation: Results are expressed as copies of pluripotency marker per copies of reference gene. Establish a clinically relevant threshold based on validation studies correlating molecular detection with functional outcomes [100].
In Vivo Tumorigenicity Assay
Principle: The gold standard for assessing teratoma formation potential involves transplantation of cells into immunocompromised mice, with monitoring for tumor development over several months [100] [103].
Reagents and Materials:
- Test article: hPSC-derived MSCs (fresh or post-thaw)
- Positive control: Parental hPSCs (1×10⁶ cells)
- Negative control: Primary MSCs from tissue sources
- Immunocompromised mice: NOD-scid IL2Rγnull (NSG) or similar
- Matrigel or similar basement membrane matrix
Procedure:
- Cell Preparation: Mix test/control cells with Matrigel (1:1 ratio) on ice to a final volume of 100-200µL per injection.
- Transplantation: Inject cells subcutaneously, intramuscularly, or into an immunologically privileged site (testis, kidney capsule) of anesthetized mice.
- Monitoring: Palpate weekly for tumor formation over 12-20 weeks, measuring dimensions with calipers.
- Termination: Euthanize animals at study endpoint or when tumors reach predetermined size limits.
- Histopathological Analysis: Excise, fix, and section tumors for hematoxylin and eosin staining, examining for characteristic teratoma features (tissues from all three germ layers).
Interpretation: A product is considered to have acceptable tumorigenicity risk if no teratomas form at the maximum clinical dose scaled by body surface area, with appropriate safety margins [100].
Integration with Cryopreservation Research
The cryopreservation process itself can impact tumorigenicity risk assessment through multiple mechanisms. Differential survival of undifferentiated versus differentiated cells during freeze-thaw cycles may alter the relative proportion of residual pluripotent cells in the final product [4] [9]. Additionally, cryopreservation-induced stress may potentially dedifferentiation or select for subpopulations with altered growth characteristics [106] [105]. Therefore, tumorigenicity assessment should be performed on both pre-freeze and post-thaw samples to fully characterize product safety.
Advanced cryopreservation strategies for tissue-engineered structures incorporating hPSC-derived MSCs present additional challenges for tumorigenicity assessment. Three-dimensional architecture and scaffold properties can influence both the survival of residual undifferentiated cells and their detection in analytical assays [9]. Furthermore, the impact of novel cryoprotectants being developed to replace DMSO—such as polyampholytes, antifreeze proteins, and synthetic polymers—on pluripotent cell viability requires careful evaluation [105] [9].
Table 2: Research Reagent Solutions for Tumorigenicity Assessment
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cell Culture Media | mTeSR1, StemFlex, Pluripotent Stem Cell SFM | Selective expansion of residual pluripotent cells in HEC assay | Defined, xeno-free formulations preferred for clinical applications |
| Cryoprotectants | DMSO, ethylene glycol, polyampholytes, PVA | Protect cells during freezing while maintaining viability | DMSO concentration and removal critical; newer agents may offer improved safety profiles |
| Molecular Assays | TaqMan ddPCR assays for OCT4, NANOG, SOX2 | Detection and quantification of pluripotency markers at high sensitivity | Requires validation for each specific hPSC line and differentiation protocol |
| Cell Separation | MACS or FACS antibodies for CD105, CD73, CD90, TRA-1-60, SSEA-4 | Enrichment of MSC population or detection of pluripotent cells | Surface marker expression may vary with culture conditions and differentiation status |
| In Vivo Models | NOD-scid IL2Rγnull (NSG) mice, Matrigel | Assessment of teratoma formation potential in permissive environment | Cost, duration, and ethical considerations; requires specialized facilities |
Data Analysis and Acceptance Criteria
Establishing scientifically justified acceptance criteria is essential for determining the safety of hPSC-derived MSC products. These criteria should be based on risk-benefit analysis considering the clinical indication, route of administration, and patient population [100] [103]. A multi-parametric approach combining results from orthogonal methods provides the most comprehensive risk assessment.
For quantitative methods like ddPCR, establish a threshold level of pluripotency marker expression below which no teratoma formation has been observed in validated animal models. The HESI International Cell Therapy Committee recommends leveraging the superior sensitivity of in vitro assays (detection limits of ≤1×10⁻⁶) over traditional in vivo methods (detection limits of ~1×10⁴ cells) for quality control purposes [100] [101].
All tumorigenicity assessment methods require thorough validation for each specific product, including determination of specificity, sensitivity, accuracy, and precision. This validation should demonstrate the ability to consistently detect residual undifferentiated cells at levels that would pose unacceptable risks in the clinical setting [100].
Workflow Integration and Decision Pathways
Implementing tumorigenicity assessment within the overall development workflow for hPSC-derived MSCs requires careful planning and strategic decision-making. The following diagram illustrates the integrated assessment strategy and critical decision points:
Integrated Tumorigenicity Assessment Workflow
This integrated approach ensures that tumorigenicity risk is assessed at critical stages of product development and manufacturing, with clear decision points guiding product progression. The workflow emphasizes the use of highly sensitive in vitro methods for routine quality control, supplemented by orthogonal methods for validation and comprehensive characterization.
Effective management of teratoma formation potential requires a systematic, multi-layered approach combining robust differentiation protocols, sensitive detection methods, and rigorous quality control. The consensus recommendation from the Health and Environmental Sciences Institute's International Cell Therapy Committee supports a transition from traditional in vivo assays to more sensitive and reproducible in vitro methods for routine quality control of hPSC-derived cell therapy products [100] [101]. Integrating these tumorigenicity assessments with cryopreservation research ensures that safety profiles are maintained throughout product storage and distribution, ultimately supporting the clinical translation of hPSC-derived MSC therapies for tissue engineering applications.
The field of regenerative medicine increasingly relies on the therapeutic potential of mesenchymal stem cells (MSCs) and, more notably, on advanced MSC-based tissue-engineered constructs. These three-dimensional (3D) systems—including cell-laden hydrogels, bioprinted structures, and tissue scaffolds—provide a more physiologically relevant environment for cells compared to traditional two-dimensional (2D) cultures, thereby enhancing their secretory activity and regenerative potential [9]. The clinical translation and widespread commercialization of these advanced therapies are critically dependent on effective long-term preservation strategies. Cryopreservation serves as a cornerstone for biobanking, enabling the on-demand availability of these biological products [9] [107]. The two predominant cryopreservation methodologies are slow freezing and vitrification, each with distinct principles, advantages, and limitations. This application note provides a comparative analysis of slow freezing versus vitrification for various MSC tissue constructs, presenting structured quantitative data, detailed experimental protocols, and key reagent solutions to inform research and development in this domain.
Comparative Analysis of Cryopreservation Modalities
The selection of a cryopreservation method significantly impacts the viability, functionality, and structural integrity of MSC constructs. The following tables summarize the core characteristics and performance outcomes of slow freezing and vitrification across different types of MSC constructs.
Table 1: Fundamental Characteristics of Slow Freezing and Vitrification
| Parameter | Slow Freezing | Vitrification |
|---|---|---|
| Basic Principle | Controlled, gradual cooling allowing cellular dehydration [108]. | Ultra-rapid cooling to form a glass-like, ice-free state [109] [108]. |
| Cooling Rate | Slow (e.g., -0.3 °C/min to -40°C) [108]. | Very rapid (requires high cooling rates) [109]. |
| CPA Mechanism | Low-to-moderate concentrations of penetrating CPAs (e.g., DMSO) [15]. | High concentrations of single or multi-component CPAs [109] [110]. |
| Primary Ice-Related Risk | Extracellular ice crystal formation [108]. | Intracellular ice formation during cooling/rewarming if conditions are not optimal [109]. |
| Primary CPA-Related Risk | Osmotic stress during addition/removal [9]. | Direct chemical toxicity and osmotic stress [109]. |
| Technical Complexity | Low; utilizes controlled-rate freezers [15]. | High; often requires specialized carriers and precise handling [110]. |
| Suitability for Complex Constructs | Established for cell suspensions and smaller scaffolds [9] [15]. | Promising for complex constructs, though sample volume is a limitation [109] [110]. |
Table 2: Performance Comparison for Different MSC Constructs
| MSC Construct Type | Cryopreservation Method | Key Findings | Reference |
|---|---|---|---|
| Ovarian Tissue (Bovine Model) | Slow Freezing vs. Vitrification | No significant difference in follicular viability post-thaw. Vitrification noted as faster, less expensive, and more adaptable to lab routine. | [111] |
| Ovarian Tissue (Human) | Slow Freezing vs. Vitrification | No significant differences in angiogenic factor secretion profiles or apoptotic indices post-culture. | [108] |
| 3D-MSCs in GelMA Hydrogel | Vitrification (with microfluidics) | 96% post-warming viability; enabled 25% reduction in required CPA concentration. Preserved mitochondrial function and wound healing capacity in vivo. | [110] |
| MSCs in PRP-SF Bioscaffold | Slow Freezing | Best results with DMSO 10% or DMSO 10% + Sucrose 0.2M. Maintained multilineage differentiation potential post-thaw. | [15] |
| MSCs in Alginate Microcapsules | Slow Freezing (Low DMSO) | Microencapsulation enabled a reduction of DMSO to 2.5% while maintaining viability above the 70% clinical threshold. | [109] |
| Adherent MSCs in Microfluidic Bioreactor | Slow Freezing | Application of low shear stress (4e-3 μbar) increased focal point adhesions (vinculin expression), enhancing cellular survivability post-thaw. | [112] |
Detailed Experimental Protocols
Protocol 1: Slow Freezing of MSCs in a PRP-SF Bioscaffold
This protocol is adapted from a study demonstrating high viability and retained differentiation potential of knee-derived MSCs cryopreserved within an allogeneic biomimetic scaffold [15].
1. Bioscaffold Preparation:
- Prepare the Platelet Rich Plasma (PRP) and Synovial Fluid (SF) mixture according to established methods [15].
- Embed MSCs within the PRP-SF solution at a desired density (e.g., ≥ 5 × 10^4 cells per construct) and activate to form the fibrin-based bioscaffold.
2. Cryoprotectant Equilibration:
- Prepare the cryopreservation medium. The optimal solutions identified are:
- Gently immerse the prepared bioscaffolds in the chosen cryoprotectant solution and incubate at 4°C for 40 minutes to allow for full penetration and equilibration [15].
3. Controlled-Rate Freezing:
- Transfer the equilibrated bioscaffolds to a controlled-rate freezer.
- Initiate the cooling program:
4. Long-Term Storage:
- Promptly transfer the frozen samples to a liquid nitrogen storage tank for long-term preservation at or below -140°C.
5. Thawing and CPA Removal:
- Rapidly thaw the bioscaffolds by placing them in a 37.2 °C water bath for approximately 130 seconds [108].
- Immediately and gradually remove the CPAs to minimize osmotic shock. This is achieved by serially washing the bioscaffolds in solutions containing decreasing concentrations of sucrose (e.g., 0.75 M, 0.375 M, 0.187 M), each for 15 minutes, followed by final washes in a CPA-free buffer [108] [15].
Protocol 2: Vitrification of 3D-MSCs Encapsulated in GelMA Hydrogel Microspheres
This advanced protocol utilizes microfluidic encapsulation and vitrification to achieve high survival with reduced CPA toxicity [110].
1. Cell Encapsulation via Microfluidics:
- Prepare a suspension of human umbilical cord MSCs (hUC-MSCs) in a sterile sodium alginate solution.
- Using a high-voltage electrostatic coaxial spraying device, generate microdroplets. The core flow (cell suspension) and shell flow (sodium alginate) are extruded at controlled rates (e.g., 25 μL/min and 75 μL/min, respectively) with an applied voltage (e.g., 6 kV) [109] [110].
- Collect the microdroplets in a calcium chloride solution to instantaneously form gelated microspheres (3D-MSCs hydrogel microspheres, 3D-MSCsHM).
- Culture the microspheres briefly to allow for cell recovery.
2. Vitrification Solution Equilibration:
3. Ultra-Rapid Cooling and Storage:
4. Rapid Warming and CPA Dilution:
- For warming, swiftly submerge the metal mesh with the samples into a pre-warmed (37°C) washing solution containing a high sucrose concentration (e.g., 0.8 M) for 1 minute [108].
- Sequentially transfer the samples to solutions with decreasing sucrose concentrations (e.g., 0.4 M for 3 minutes) and finally to a basal culture medium to completely remove the CPAs [108] [110].
Workflow and Decision-Making Diagram
The following diagram illustrates the key decision points and experimental workflows for selecting and implementing the two cryopreservation methods for MSC constructs.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Cryopreservation of MSC Constructs
| Reagent/Material | Function/Description | Example Application |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant; prevents intracellular ice formation by binding water and protecting membrane structures [9]. | Standard CPA in slow freezing (e.g., 10% for PRP-SF scaffolds) [15]. |
| Ethylene Glycol (EG) | Penetrating cryoprotectant; lower molecular weight can facilitate faster penetration into tissues [9] [108]. | Component of vitrification solutions for ovarian tissue [108]. |
| Sucrose | Non-penetrating cryoprotectant; acts as an osmotic buffer, countering salt concentration effects and reducing required penetrating CPA concentration [9] [109]. | Used in both slow freezing (0.2M) and vitrification (0.5-0.8M) as a component of freezing/thawing solutions [109] [108] [15]. |
| Sodium Alginate | Natural biomaterial for forming hydrogel microcapsules; provides a 3D cryoprotective environment for encapsulated cells [109]. | Microencapsulation of MSCs for low-DMSO cryopreservation [109]. |
| GelMA (Gelatin Methacryloyl) | Photocrosslinkable hydrogel; provides a tunable 3D scaffold that mimics the extracellular matrix, enhancing post-thaw viability and function [110]. | Vitrification of 3D-MSCs for wound healing applications [110]. |
| Polyvinylpyrrolidone (PVP) | Synthetic polymer; non-penetrating CPA that increases solution viscosity, inhibiting ice crystal growth [9] [108]. | Additive in vitrification solutions (e.g., 5%) [108]. |
| Platelet Rich Plasma (PRP) & Synovial Fluid (SF) | Components of an allogeneic, biomimetic bioscaffold; provides a natural fibrin structure and native growth factors for embedded MSCs [15]. | Cryopreservation of knee-derived MSCs within a native-like environment [15]. |
| Customized Metal Meshes | Vitrification carrier; provides a large surface area for ultra-rapid heat transfer during plunging into liquid nitrogen [108]. | Vitrification of ovarian tissue strips and hydrogel microspheres [108] [110]. |
Both slow freezing and vitrification are viable methods for the cryopreservation of MSC-based tissue constructs, with the optimal choice being highly dependent on the specific construct's properties and the intended clinical or research application. Slow freezing, characterized by its use of lower CPA concentrations and simpler equipment, remains a robust and widely applicable method, particularly for cell suspensions and larger scaffold-based constructs. Vitrification, while technically more demanding and requiring high cooling rates, offers a significant advantage by eliminating ice crystal formation and is showing great promise for complex micro-tissue constructs like hydrogel-encapsulated MSCs, especially when combined with technologies like microfluidics. The ongoing development of advanced biomaterials and optimized CPA cocktails is consistently improving post-thaw outcomes, facilitating the path toward readily available, off-the-shelf regenerative therapies. This comparative analysis provides a foundational framework and practical tools for researchers to advance the crucial field of biobanking for tissue engineering.
This document outlines the essential biosafety and regulatory protocols for researching cryopreserved Mesenchymal Stem Cell (MSC)-based tissue-engineered structures (TES). Ensuring the safety, identity, and purity of these advanced therapy medicinal products (ATMPs) is paramount for their clinical translation. This document provides detailed application notes and standardized protocols for donor eligibility screening, cell line authentication, and process validation, tailored specifically for the development of MSC-based TES under Good Manufacturing Practice (GMP)-compliant conditions [113]. Adherence to these guidelines, such as those from the International Society for Stem Cell Research (ISSCR) and the U.S. Food and Drug Administration (FDA), is not only a regulatory expectation but a fundamental requirement to ensure the integrity of research and the safety of future patients [114] [115].
Regulatory Framework and Fundamental Principles
The development of MSC-based TES must be guided by a framework of ethical and regulatory principles that underpin all research activities.
- Integrity of the Research Enterprise: Research must be designed to ensure information is trustworthy and reliable, maintained through independent peer review, oversight, and accountability at every stage [114].
- Primacy of Patient Welfare: The duty of care to patients and research subjects is paramount. This includes protecting vulnerable patients from excessive risk and ensuring unproven interventions are not marketed outside of formal, regulated research settings [114].
- Transparency and Social Justice: Researchers should promote the timely sharing of data and communicate accurately with the public. The benefits and burdens of clinical translation should be distributed justly, with an emphasis on addressing unmet medical needs and enrolling diverse populations in clinical trials [114].
Regulatory oversight for such products often falls under the FDA's regulations for Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) [115]. Establishments must comply with donor eligibility requirements outlined in 21 CFR Part 1271, Subpart C, which form the basis for the donor screening protocol in Section 3 [115].
Donor Screening and Eligibility Determination
Rigorous donor screening is the first critical step in mitigating the risk of transmitting communicable diseases.
Application Notes
Donor eligibility determination is mandatory for all donors of HCT/Ps. The objective is to identify donors who may present an increased risk of transmitting relevant communicable diseases to recipients [115]. This process involves a donor medical history interview and specific communicable disease testing.
Protocol: Donor Eligibility Determination
Objective: To establish standardized procedures for screening and testing potential donors of MSC tissue for TES, ensuring compliance with regulatory requirements [115].
Materials and Reagents:
- Donor History Questionnaire (DHQ)
- FDA-licensed, approved, or cleared donor screening tests
- Specimen collection kits (for blood samples)
Workflow:
- Donor Identification and Consent: Obtain informed consent from the potential donor or their legally authorized representative.
- Medical History Interview: Conduct a complete medical history interview using a standardized DHQ to identify risk factors for communicable diseases.
- Donor Physical Assessment: Perform a physical assessment to check for signs of infection or disease.
- Blood Collection: Collect a blood sample from the donor using an aseptic technique.
- Communicable Disease Testing: Test the donor sample using approved methods for the following agents (as recommended by the FDA [115]):
- Human Immunodeficiency Virus (HIV-1 and HIV-2)
- Hepatitis B Virus (HBV)
- Hepatitis C Virus (HCV)
- Treponema pallidum (Syphilis)
- Chlamydia trachomatis and Neisseria gonorrhoeae
- Human T-Lymphotropic Virus (HTLV)
- Cytomegalovirus (CMV)
- Mycobacterium tuberculosis (Mtb)
- Eligibility Determination: Review all collected data. A donor is considered eligible only if the medical history, physical assessment, and test results do not indicate a risk of communicable disease transmission.
The following diagram illustrates the sequential steps for determining donor eligibility.
Cell Line Authentication and Characterization
Preventing cross-contamination or misidentification of cell lines is crucial for research reproducibility and product safety.
Application Notes
Short Tandem Repeat (STR) profiling is the gold standard method for authenticating human cell lines [116]. Next-Generation Sequencing (NGS)-based STR profiling (STR-NGS) offers advantages over traditional capillary electrophoresis (STR-CE), including the ability to discern nucleotide variations within STR loci, higher sensitivity, and flexible multiplexing, which is valuable for detecting cross-contamination in mixed samples [116]. For MSCs, authentication must be coupled with phenotypic and functional characterization to confirm "stemness" and functionality pre- and post-cryopreservation.
Protocol: STR Profiling via Next-Generation Sequencing
Objective: To authenticate human MSC lines using high-throughput STR-NGS, confirming unique genetic identity and detecting potential contamination [116].
Materials and Reagents:
- Genomic DNA (70-140 ng) from MSC sample [116]
- PCR1 Master Mix: Platinum SuperFi II PCR Master Mix, locus-specific primers, TMAO additive [116]
- PCR2 Master Mix: High-fidelity polymerase, dual indexing adaptors [116]
- Illumina MiSeq System (or equivalent NGS platform) [116]
- Bioinformatics Tool: STRight Python program or equivalent [116]
Workflow:
- DNA Extraction: Isolate high-quality genomic DNA from the MSC sample.
- Library Preparation (PCR1): Amplify STR loci of interest (e.g., CODIS loci) in a multiplexed PCR reaction using the optimized SuperFi+TMAO master mix. This step adds partial Illumina adapters [116].
- Indexing (PCR2): Perform a second, limited-cycle PCR to add unique dual indices and complete adapter sequences for multiplexed sequencing [116].
- Sequencing: Pool indexed libraries and sequence on an Illumina MiSeq platform.
- Data Analysis: Use the STRight program to analyze sequencing data, call alleles based on repeat number and sequence, and generate a profile [116].
- Interpretation: Compare the resulting STR profile to reference databases (e.g., ATCC) or original donor tissue to confirm identity. Investigate any additional allele peaks that may indicate contamination.
Characterization of Mesenchymal Stem Cells
Beyond STR profiling, MSCs must be characterized for identity and function, especially after cryopreservation. Key parameters to assess are summarized in the table below.
Table 1: Key Characterization Parameters for Cryopreserved MSCs
| Parameter | Assay/Method | Acceptance Criteria for MSCs | Purpose |
|---|---|---|---|
| Cell Viability | Live-Dead Staining (Calcein AM/ Ethidium homodimer) or Annexin V-PI [117] | >70-80% post-thaw viability [118] [117] | Measures immediate cryopreservation damage and cell death. |
| Cell Phenotype | Flow Cytometry [117] | Positive for: CD73, CD90, CD105Negative for: CD34, CD45, CD14, CD19 [117] | Confirms MSC immunophenotype and purity. |
| Differentiation Potential | Trilineage Differentiation & Staining [117] | Adipogenesis: Lipid vesicles (Oil Red O)Osteogenesis: Calcium deposits (Alizarin Red)Chondrogenesis: Glycosaminoglycans (Alcian Blue) [117] | Verifies functional multipotency post-thaw. |
| Immunomodulatory Ability | Co-culture with PBMCs & T-cell proliferation/cytokine assays [117] | Suppression of T-cell proliferation [117] | Confirms retention of key therapeutic function. |
Process Validation and Quality Control
A validated and controlled cryopreservation process is essential to ensure the final TES product retains its critical quality attributes.
Application Notes
Process validation provides documented evidence that the cryopreservation process consistently produces a TES meeting its predetermined specifications and quality attributes. This involves defining and controlling Critical Process Parameters (CPPs) like cooling rate and storage temperature, and monitoring Critical Quality Attributes (CQAs) like viability and functionality (as in Table 1) [117] [113]. Quality control extends to selecting GMP-grade, xeno-free reagents to ensure patient safety and regulatory compliance [117].
Protocol: Validation of the Cryopreservation Process for MSC-Based Constructs
Objective: To establish and validate a controlled-rate freezing and storage protocol for MSC-seeded scaffolds, ensuring consistent post-thaw viability and functionality.
Materials and Reagents:
- Cryoprotective Agent (CPA): e.g., 10% DMSO in xeno-free cryomedium or DMSO-free alternatives like CryoOx [113] [34]. For some constructs, 10% Ethylene Glycol may also be suitable [118].
- Cryobags or Cryovials [118]
- Controlled-Rate Freezer (or an alcohol-free freezing container achieving -1°C/min) [117]
- Liquid Nitrogen Storage Tank (-196°C) or -150°C ultra-low freezer [117]
- Water Bath (37°C)
Workflow:
- Pre-freeze Analysis: Confirm cell viability, phenotype, and scaffold properties before freezing.
- CPA Addition: Gently aspirate culture medium from the TES and add pre-chilled CPA. Use a stepwise addition if required to minimize osmotic shock.
- Packaging: Aseptically transfer the TES in CPA into a final container (e.g., cryobag). Cryobags can improve heat transfer compared to multiwell plates [118].
- Controlled-Rate Freezing: Place samples in a controlled-rate freezer. Apply a standard cooling rate of -1°C/min until the target temperature (e.g., -80°C to -100°C) is reached [117] [34].
- Long-Term Storage: Transfer frozen samples to a long-term storage unit (liquid nitrogen vapor phase, -150°C to -196°C, is optimal) [117].
- Thawing and Post-Thaw Processing: Rapidly thaw samples in a 37°C water bath with gentle agitation until just ice-free. Immediately dilute the CPA with pre-warmed culture medium and centrifuge to remove the CPA [117].
- Post-Thaw Analysis: Assess the CQAs as defined in Table 1 (viability, phenotype, functionality) and compare to pre-freeze and control values.
The interaction of process parameters, quality attributes, and regulatory guidelines is summarized in the following diagram.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Reagent Solutions for MSC-based TES Cryopreservation Research
| Item | Function/Application | Key Considerations |
|---|---|---|
| Xeno-Free Cryomedium | Formulates CPA solution for clinical-grade applications. | Must be chemically defined, animal-origin-free to prevent immunogenic reactions and ensure lot-to-lot consistency [117]. |
| DMSO & DMSO-Free CPAs | Permeating CPA to protect cells from ice crystal formation. | 10% DMSO is common but can be cytotoxic. DMSO-free alternatives (e.g., CryoOx) are emerging to improve safety profiles [113] [34]. |
| Polymer-Based NPAs (e.g., Trehalose, Sucrose) | Non-permeating CPA that acts extracellularly. | Often used in vitrification mixtures with lower DMSO concentrations to reduce toxicity and osmotic shock [34]. |
| Platinum SuperFi DNA Polymerase | High-fidelity enzyme for STR-NGS library amplification. | Provides high yield and accuracy for multiplex PCR1 of STR loci, crucial for reliable sequencing [116]. |
| Viability & Apoptosis Assays (e.g., Calcein AM/EtD-1, Annexin V-PI) | Quantifies live/dead cells and detects early apoptosis post-thaw. | Critical for assessing immediate cryopreservation injury and cell quality [117]. |
| Flow Cytometry Antibody Panels | Confirms MSC immunophenotype (CD73+/CD90+/CD105+; CD34-/CD45-/CD14-). | Essential for identity and purity testing before and after cryopreservation [117]. |
| Trilineage Differentiation Kits | Functional validation of MSC multipotency post-thaw. | Kits typically include induction media and stains (Oil Red O, Alizarin Red, Alcian Blue) [117]. |
| GMP-Grade Bioreactors/Scaffolds | Provides the 3D structural component for the TES. | Materials (e.g., electrospun PCL/PLA) must be biocompatible and suitable for GMP manufacturing [118] [113]. |
Mesenchymal stem cells (MSCs) have emerged as a highly promising strategy in regenerative medicine due to their self-renewal, pluripotency, and immunomodulatory properties [1]. These non-hematopoietic, multipotent stem cells can differentiate into various mesodermal lineages and modulate the immune system through both direct cell–cell interactions and the release of immunoregulatory molecules [1]. The therapeutic potential of MSCs from different tissues has been widely explored in preclinical models and clinical trials for a spectrum of human diseases, positioning them as a cornerstone of advanced tissue-engineered therapies [1].
The transition of MSC-based therapies from research to clinical application is intrinsically linked to advances in cryopreservation techniques, which enable the long-term storage and stability required for off-the-shelf availability and standardized treatment protocols. Effective cryopreservation maintains MSC functionality and viability, making it a critical step in the manufacturing and clinical deployment of tissue-engineered structures [4]. This article examines the clinical trial evidence for MSC applications in hematological diseases, orthopedic repair, and diabetes, with a specific focus on the experimental protocols and cryopreservation context essential for research and drug development.
Clinical Applications and Trial Evidence
The therapeutic application of MSCs spans diverse medical fields. The table below summarizes the clinical trial evidence for their use in hematological, orthopedic, and diabetic conditions.
Table 1: Clinical Trial Evidence for MSC-Based Therapies
| Disease Area | Therapeutic Mechanism | Reported Efficacy in Clinical Trials | Key Challenges |
|---|---|---|---|
| Hematological Diseases (e.g., Graft-versus-Host Disease) | Immunomodulation via T-cell suppression, interaction with dendritic cells, and release of anti-inflammatory factors [1]. | Demonstrated efficacy in modulating immune responses; over 2,300 human clinical trials involving MSCs registered [119]. | Infusion-related toxicity, donor variability, and functional heterogeneity of cells [119]. |
| Orthopedic Repair (e.g., Bone Fractures, Osteoarthritis) | Differentiation into osteoblasts and chondrocytes; paracrine secretion of bone morphogenetic proteins (BMPs), VEGF, and extracellular vesicles to promote angiogenesis and matrix remodeling [120]. | Early-phase trials report promoted bone regeneration, reduced pain, and decreased need for autologous grafts [120]. One completed trial for osteoarthritis in Italy [119]. | Poor cell survival and integration post-transplantation; inconsistent osteogenic potential [120]. |
| Diabetes (Type 1 & 2) | Immunomodulation to protect pancreatic β-cells; differentiation into insulin-producing cells; secretion of trophic factors to improve insulin sensitivity [121] [122]. | Modest improvements in HbA1c and reduced exogenous insulin requirements in early-phase trials [121]. Preservation of β-cell mass and improved islet graft acceptance in T1D [122]. | Small, heterogeneous studies with short follow-up; impaired cell function in the diabetic microenvironment [121]. |
Experimental Protocols for MSC Processing and Evaluation
Protocol: Isolation and Expansion of Adipose-Derived MSCs (AD-MSCs)
Application Note: This protocol is foundational for generating a cellular product for research and potential clinical use, particularly in diabetes and orthopedic repair, given the high yield and potent immunomodulatory effects of AD-MSCs [122].
- Tissue Harvesting: Obtain subcutaneous adipose tissue from surgical waste (e.g., lipoaspirates) under informed consent and ethical approval.
- Enzymatic Digestion: Mince the tissue and digest with 0.05–0.15% collagenase Type I or II for 30–90 minutes at 37°C with constant agitation [122].
- Stromal Vascular Fraction (SVF) Isolation: Centrifuge the digest to separate the buoyant adipocytes from the pelleted SVF. Lyse red blood cells and filter the SVF through 70–250 μm nylon mesh to remove debris [122].
- Plastic Adherence & Culture: Seed the SVF cells in culture flasks with a standard growth medium (e.g., DMEM/F12 supplemented with fetal bovine serum). Incubate at 37°C with 5% CO₂ [122].
- Cell Passaging: Remove non-adherent cells after 24-48 hours by washing. Allow AD-MSCs to expand until 70-80% confluency, then passage using trypsin/EDTA.
- Phenotypic Validation: Characterize cells via flow cytometry. AD-MSCs must be ≥95% positive for CD73, CD90, and CD105, and ≤2% positive for CD45, CD34, CD14 or CD11b, CD79α or CD19, and HLA-DR [1] [122].
Protocol: Cryopreservation and Thawing of MSCs
Application Note: This slow-freezing protocol is critical for creating biobanks of MSCs for tissue-engineered structures, ensuring consistent quality and functionality for future clinical applications [4].
- Pre-Freezing Preparation: Harvest MSCs at the desired passage (e.g., P3-P5) and resuspend them in a cryopreservation medium at a concentration of 1-5 x 10^6 cells/mL. A typical medium consists of 90% FBS and 10% DMSO, though serum-free and lower DMSO alternatives are being explored [4].
- Slow Freezing Process:
- Aliquot the cell suspension into cryogenic vials.
- Place vials in a controlled-rate freezer or a -80°C freezer using an isopropanol freezing container to achieve an approximate cooling rate of -1°C to -3°C per minute [4].
- After 24 hours at -80°C, transfer the vials to the vapor or liquid phase of liquid nitrogen (-135°C to -196°C) for long-term storage.
- Thawing and Post-Thaw Processing:
- Rapidly thaw cryopreserved vials in a 37°C water bath until only a small ice crystal remains (≈1-2 minutes) [4].
- Decontaminate the vial with 70% ethanol before opening.
- Gently transfer the cell suspension to a centrifuge tube prefilled with pre-warmed culture medium to dilute the DMSO.
- Centrifuge at 300-400 x g for 5-10 minutes to pellet the cells and remove the cryoprotectant-containing supernatant.
- Resuspend the cell pellet in fresh culture medium and plate for subsequent experiments. Assess viability, typically expected to be 70-80% [4].
Protocol: In Vitro Osteogenic Differentiation Assay
Application Note: This protocol is used to validate the bone-forming potential of MSCs, particularly for orthopedic applications, and to test the efficacy of functionalized or cryopreserved cells [120].
- Cell Seeding: Seed MSCs (e.g., BM-MSCs or AD-MSCs) at a density of 5,000-10,000 cells/cm² in culture plates.
- Osteogenic Induction: Once cells reach 60-70% confluency, replace the growth medium with osteogenic induction medium. This typically consists of basal medium supplemented with 50 µM ascorbate-2-phosphate, 10 mM β-glycerophosphate, and 100 nM dexamethasone [120].
- Maintenance: Culture the cells for 21-28 days, changing the induction medium every 2-3 days.
- Endpoint Analysis:
- Alizarin Red S Staining: Fix cells with 4% paraformaldehyde and stain with 2% Alizarin Red S (pH 4.1-4.3) to detect calcium-rich mineralized nodules.
- Alkaline Phosphatase (ALP) Activity: Measure ALP activity using a colorimetric assay or specific stain as an early marker of osteogenic differentiation.
- Gene Expression Analysis: Use qRT-PCR to analyze the upregulation of osteogenic genes such as RUNX2 (master transcription factor), osteocalcin (OCN), and osteopontin (OPN) [120].
Signaling Pathways in MSC-Mediated Therapy
The therapeutic effects of MSCs are mediated through complex signaling pathways that direct their differentiation and immunomodulatory functions. The following diagrams illustrate key pathways in bone repair and immunomodulation.
Wnt/β-catenin Pathway in Osteogenesis
This pathway is crucial for directing MSCs toward bone-forming osteoblasts, a key mechanism for orthopedic repair [120].
MSC Immunomodulation of T-cell Response
This pathway depicts how MSCs exert immunomodulatory effects, which is central to their application in hematological diseases and autoimmune conditions like diabetes [1] [122].
The Scientist's Toolkit: Research Reagent Solutions
Successful MSC research and therapy development rely on a suite of critical reagents and materials. The following table details key components and their functions.
Table 2: Essential Research Reagents for MSC-Based Therapy Development
| Reagent/Material | Function/Application | Examples & Notes |
|---|---|---|
| Cryoprotective Agents (CPAs) | Protect cells from ice crystal damage during freeze-thaw cycles [4]. | DMSO (standard, but has toxicity concerns [123]), Trehalose (non-permeating, safer alternative). |
| Cell Surface Markers | Identification and phenotypic validation of MSCs via flow cytometry [1] [122]. | Positive Panel: CD73, CD90, CD105. Negative Panel: CD45, CD34, HLA-DR. |
| Osteogenic Induction Cocktail | Directs MSC differentiation into osteoblasts for bone repair studies [120]. | Typically contains Dexamethasone, β-Glycerophosphate, and Ascorbic Acid. |
| Enzymatic Isolation Kits | Isolation of MSCs from tissue sources (e.g., adipose, bone marrow) [122]. | Collagenase blends for digesting adipose tissue to obtain the Stromal Vascular Fraction (SVF). |
| 3D Scaffolds/Biomaterials | Provide structural support for MSCs in tissue-engineered constructs for orthopedic repair [120]. | Injectable hydrogels, 3D-printed scaffolds made from biocompatible polymers (e.g., PLGA, chitosan). |
Clinical evidence continues to demonstrate the significant potential of MSC therapies across hematological, orthopedic, and metabolic diseases. The successful translation of this potential into reliable treatments is inextricably linked to robust cryopreservation protocols that ensure cell viability, functionality, and off-the-shelf availability for tissue-engineered structures. Future progress hinges on addressing key challenges such as donor variability, functional heterogeneity, and the development of safer, defined cryoprotectant solutions [121] [4]. The integration of advanced technologies—including bioengineering, genetic modification of MSCs, and the use of MSC-derived extracellular vesicles—is poised to enhance therapeutic efficacy and pave the way for next-generation, precision regenerative medicines [119] [120].
Conclusion
The successful cryopreservation of MSC-based tissue-engineered structures is not merely a logistical step but a critical determinant of clinical efficacy. This synthesis confirms that while established slow-freezing methods provide a reliable foundation, significant challenges remain in optimizing cryoprotectant regimens and ensuring consistent post-thaw functionality. The future of the field hinges on developing novel technological approaches—including DMSO-free cryoprotectant solutions and advanced vitrification techniques—that can better preserve both cellular viability and complex structural integrity. Furthermore, standardized validation frameworks and comprehensive biosafety assessments are urgently needed to bridge the gap between laboratory innovation and clinical application. As cryopreservation protocols evolve, they will unlock the full potential of off-the-shelf MSC products, ultimately advancing the treatment of degenerative diseases, genetic disorders, and tissue damage through reproducible, safe, and effective regenerative therapies.
References
- 1. Mesenchymal stem cells in treating human diseases: molecular mechanisms and clinical studies | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-025-02313-9]
- 2. Cell surface markers for mesenchymal stem cells related to ... [https://www.sciencedirect.com/science/article/pii/S2405844023006710]
- 3. Validated methods for isolation and qualification of ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06972-8]
- 4. Effects, methods and limits of the cryopreservation on ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-03954-3]
- 5. Mesenchymal stem cells: opening a new chapter in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465317/]
- 6. In Vitro Cell Surface Marker Expression on Mesenchymal ... [https://link.springer.com/article/10.1007/s12015-024-10743-1]
- 7. Understanding the molecular basis of mesenchymal stem ... [https://www.nature.com/articles/s41419-025-08094-x]
- 8. Mesenchymal stem cell mechanobiology and emerging experimental platforms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3673151/]
- 9. Cryostorage of Mesenchymal Stem Cells and Biomedical Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9454587/]
- 10. Comparative Analysis of Human Mesenchymal Stem Cells ... [https://www.mdpi.com/1422-0067/14/9/17986]
- 11. Comparison of infant bone marrow- and umbilical cord ... [https://www.sciencedirect.com/science/article/pii/S235232042400172X]
- 12. Comparative analysis of mesenchymal stem cells derived from ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-018-1104-x]
- 13. International Journal of Molecular Medicine [https://www.spandidos-publications.com/10.3892/ijmm.2015.2413]
- 14. Comparison of human mesenchymal stem cells derived ... [https://pubmed.ncbi.nlm.nih.gov/23318344/]
- 15. Cryopreservation of Human Mesenchymal Stem Cells in an ... [https://www.nature.com/articles/s41598-017-16134-6]
- 16. Optimizing cryopreservation strategies for scalable cell therapies [https://pmc.ncbi.nlm.nih.gov/articles/PMC11659802/]
- 17. Current challenges and future directions of ATMPs in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12274769/]
- 18. Specifics of Cryopreservation of Hydrogel Biopolymer ... [https://www.mdpi.com/2073-4360/16/2/247]
- 19. Mesenchymal Stem Cell Immunomodulation: Mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7751844/]
- 20. Deep technology for the optimization of cryostorage - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10371920/]
- 21. Overcoming ice: cutting-edge materials and advanced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11887326/]
- 22. Encapsulation for efficient cryopreservation [https://www.degruyterbrill.com/document/doi/10.1515/fzm-2025-0008/html?srsltid=AfmBOoralXHof4dZ8zbGRvTd0OZrClOuLDzc3AkrJ0X7boRAVQj3Rg7U]
- 23. Winter is coming: the future of cryopreservation - BMC Biology [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-021-00976-8]
- 24. Modeling and typical cases analyze at the cell-scale of ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025000161]
- 25. Automated Manufacturing Processes and Platforms for ... [https://link.springer.com/article/10.1007/s12015-024-10812-5]
- 26. Cryopreservation of mesenchymal stem/stromal cells using ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324924007734]
- 27. Effects, methods and limits of the cryopreservation on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11441116/]
- 28. Cryopreserved mesenchymal stem cells regain functional ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-019-2038-5]
- 29. Short-Term Cryopreservation Preserved the Function of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12524150/]
- 30. Cryostorage of Mesenchymal Stem Cells and Biomedical ... [https://www.mdpi.com/2073-4409/11/17/2691]
- 31. Advances in Cryopreservation Strategies for 3D Biofabricated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295925/]
- 32. Slow freezing [https://bio-protocol.org/exchange/minidetail?id=19079401&type=30]
- 33. Cryopreservation of biological materials: applications and ... [https://link.springer.com/article/10.1007/s11626-025-01027-0]
- 34. Cryopreservation: An Overview of Principles and Cell- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7995302/]
- 35. Development of additive materials for spheroid ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925002981]
- 36. Cryopreservation of tissues by slow-freezing using an ... [https://www.nature.com/articles/s41598-022-23913-3]
- 37. Technologies for Vitrification Based Cryopreservation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10215456/]
- 38. Cryopreservation by Directional Freezing and Vitrification ... [https://www.mdpi.com/2073-4409/11/7/1072]
- 39. Droplet based vitrification for cell aggregates: Numerical ... [https://www.sciencedirect.com/science/article/abs/pii/S1751616118303850]
- 40. Higher glass transition temperatures reduce thermal stress ... [https://www.nature.com/articles/s41598-025-13295-7]
- 41. A non-traditional approach to cryopreservation by ultra ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6645672/]
- 42. Cryopreservation and its clinical applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5395684/]
- 43. Trehalose in cryopreservation. Applications, mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11411628/]
- 44. Proteomic approach for evaluating cryoprotectant ... [https://www.nature.com/articles/s41598-025-00534-0]
- 45. The combination of trehalose and glycerol: an effective and ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-020-01969-0]
- 46. Impact of Cryopreservation and Freeze-Thawing on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9045030/]
- 47. Requirements for standardizing the assessment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11924026/]
- 48. The Impact of Varying Cooling and Thawing Rates on ... [https://www.nature.com/articles/s41598-019-39957-x]
- 49. Development of a 3-in-1 multifunctional cell processing ... [https://www.sciencedirect.com/science/article/abs/pii/S0735193324012739]
- 50. Theoretical optimization of the removal of cryoprotective ... [https://biomedical-engineering-online.biomedcentral.com/articles/10.1186/1475-925X-13-120]
- 51. Cell Cryopreservation: Common Protocols, Thawing Tips ... [https://www.genfollower.com/cell-cryopreservation-thawing-guide/]
- 52. Cryopreservation of mammalian cell lines video protocol [https://www.abcam.com/en-us/technical-resources/protocols/cryopreservation-of-mammalian-cell-lines]
- 53. Cryopreserved Vials Storage and Thawing Protocol [https://allcells.com/resources-and-support/protocols/storage-thawing/]
- 54. Cryopreservation and post-thaw differentiation of monocytes ... [https://pubs.rsc.org/en/content/articlehtml/2025/lp/d5lp00131e]
- 55. Protocols and Best Practices for Freezing Cells [https://www.stemcell.com/cryopreservation-basics-protocols-and-best-practices-for-freezing-cells]
- 56. Impact of pre- and post-thaw processing on recovery ... [https://pubmed.ncbi.nlm.nih.gov/41026081/]
- 57. Impact of pre- and post-thaw processing on recovery ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925008400]
- 58. Cryopreservation of Cell/Scaffold Tissue-Engineered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3483048/]
- 59. Hyaluronic acid incorporation into hybrid alginate-based ... [https://www.sciencedirect.com/science/article/pii/S1773224725001522]
- 60. Development of scaffold-free tissue-engineered constructs ... [https://pubmed.ncbi.nlm.nih.gov/39188648/]
- 61. Scaffold-free human mesenchymal stem cell construct ... [https://www.nature.com/articles/s42003-020-01576-y]
- 62. Advancing Scaffold Architecture for Bone Tissue Engineering [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470323/]
- 63. Overcoming ice: cutting-edge materials and advanced ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03265-6]
- 64. Cryoprotectant Toxicity: Facts, Issues, and Questions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4620521/]
- 65. Dimethyl sulfoxide in cryopreserved mesenchymal stromal cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12362920/]
- 66. A bioinspired and chemically defined alternative to dimethyl ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8563414/]
- 67. Cryopreserved clumps of mesenchymal stem cell/extracellular ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-018-0826-0]
- 68. 138 Mesenchymal Stem/Stromal Cells XT-THRIVE, A NON- ... [https://www.sciencedirect.com/science/article/pii/S1465324925002026]
- 69. Freezing cells for success: DMSO-free redefining the future ... [https://www.news-medical.net/news/20251125/Freezing-cells-for-success-DMSO-free-redefining-the-future-of-cryopreservation.aspx]
- 70. Comparison of the Effects of Different Cryoprotectants on Stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4685149/]
- 71. Effect of cooling rate and cryoprotectant concentration on ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224013001211]
- 72. Ice formation and its elimination in cryopreservation of ... [https://www.nature.com/articles/s41598-024-69528-8]
- 73. Advanced technologies for the preservation of mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8765766/]
- 74. Cryopreservation - Key Survey Findings from the ISCT ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/05/28/cryopreservation-key-survey-findings-from-the-isct]
- 75. Reconstitution and post-thaw storage of cryopreserved ... [https://www.sciencedirect.com/science/article/pii/S2352320423000275]
- 76. Clinical experience with cryopreserved mesenchymal stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11947892/]
- 77. Optimization of UC-MSCs cold-chain storage by minimizing ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224020306544]
- 78. How Contamination Happens in Labs (and How to Prevent It) [https://www.msesupplies.com/blogs/news/how-contamination-happens-in-labs-and-how-to-prevent-it?srsltid=AfmBOootYgTrGoCQxUOglWFQ7XrLT4uSaBfyGnx5ZCyJQHhlrH6qlVKF]
- 79. Cryopreservation as a versatile strategy for the construction ... [https://www.degruyterbrill.com/document/doi/10.1515/fzm-2025-0013/html?srsltid=AfmBOorRkseXlPpFOtTVFPYBMsOvsGUndwNIBEg4jHyTnBqsrWFeSFpo]
- 80. Microbiological risk assessment during the procurement ... [https://www.nature.com/articles/s41598-025-24496-5]
- 81. CellShield™ MSC Cryo Kit [https://eviabio.com/product/cellshield-msc/]
- 82. What are Safety Protocols in Handling Frozen Cells? [https://www.coleparmer.com/blog/safety-protocols-for-handling-frozen-cells/]
- 83. of Human Cryopreservation for Clinical... Mesenchymal Stem Cells [https://pubmed.ncbi.nlm.nih.gov/26280501/]
- 84. Frontiers | Key quality parameter comparison of mesenchymal ... stem [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1412811/full]
- 85. Immunomodulatory Functions of Mesenchymal Stem Cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12572674/]
- 86. Evaluation of the viability and functionality of human ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1627973/full]
- 87. Comparative Analysis of Serum and Serum-Free Medium ... [https://www.mdpi.com/1422-0067/25/19/10627]
- 88. Development of scaffold-free tissue-engineered constructs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11344744/]
- 89. Automated Manufacturing Processes and Platforms for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11872983/]
- 90. Comparison of culture media supplements identifies serum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382287/]
- 91. Optimizing mesenchymal stem cell therapy: from isolation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12054297/]
- 92. Optimization of Mesenchymal Stromal Cell (MSC ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.918565/full]
- 93. Impact of Cryopreservation and Freeze-Thawing on ... [https://link.springer.com/article/10.1007/s40778-022-00212-1]
- 94. Mesenchymal stromal cells derived from various tissues [https://www.sciencedirect.com/science/article/pii/S0011224015002047]
- 95. Cryopreserved mesenchymal stem cells regain functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6716839/]
- 96. Cryopreserved hMSCs Maintain Comparable in vitro ... [https://www.roosterbio.com/blog/cryopreserved-hmscs-maintain-comparable-in-vitro-functional-activity-compared-to-fresh-hmscs/]
- 97. Optimal Sca-1-based procedure for purifying mouse adipose ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12122437/]
- 98. Chondrogenic and Osteogenic In Vitro Differentiation ... [https://www.mdpi.com/2077-0383/14/4/1184]
- 99. Optimization of Human Adipose-Derived Mesenchymal ... [https://bmrat.org/index.php/BMRAT/article/view/988]
- 100. Evaluating teratoma formation risk of pluripotent stem cell ... [https://pubmed.ncbi.nlm.nih.gov/40392167/]
- 101. Evaluating teratoma formation risk of pluripotent stem cell- ... [https://hesiglobal.org/publication/evaluating-teratoma-formation-risk-of-pluripotent-stem-cell-derived-cell-therapy-products-a-consensus-recommendation-from-the-health-and-environmental-sciences-institutes-international-cell/]
- 102. Evaluating teratoma formation risk of pluripotent stem cell- ... [https://oak.novartis.com/55827/]
- 103. Risk factors in the development of stem cell therapy [https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-9-29]
- 104. The Evolving Landscape of Stem Cell Therapies for ... [https://link.springer.com/article/10.1007/s40291-025-00816-3]
- 105. Advanced cryopreservation engineering strategies: the critical ... [https://cellregeneration.springeropen.com/articles/10.1186/s13619-023-00173-8]
- 106. Genetic Stability of Mesenchymal Stromal Cells for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6566307/]
- 107. Biofabrication & cryopreservation of tissue engineered ... [https://pubmed.ncbi.nlm.nih.gov/39258414/]
- 108. of angiogenic potential in Comparison . vitrified ... vs slow frozen [https://www.nature.com/articles/s41598-023-39920-x?error=cookies_not_supported&code=d2c223e2-7d50-4f0c-ac78-090198c43b16]
- 109. Hydrogel microcapsulation technology reduces the DMSO ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12639263/]
- 110. Vitrification of 3D-MSCs encapsulated in GelMA hydrogel [https://www.sciencedirect.com/science/article/pii/S014181302500265X]
- 111. between Comparison and Slow in Terms of... Freezing Vitrification [https://pubmed.ncbi.nlm.nih.gov/27472810/]
- 112. Enhanced cryopreservation of MSCs in microfluidic ... [https://www.nature.com/articles/srep35416]
- 113. Reassessing Long-Term Cryopreservation Strategies for ... [https://www.sciencedirect.com/science/article/pii/S0022202X24018980]
- 114. Guidelines — International Society for Stem Cell Research [https://www.isscr.org/guidelines]
- 115. Recommendations for Determining Eligibility of Donors ... [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-determining-eligibility-donors-human-cells-tissues-and-cellular-and-tissue-based]
- 116. Short tandem repeat profiling via next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618599/]
- 117. Dealing with Cryopreservation Challenges in ... [https://www.atlantisbioscience.com/blog/dealing-with-cryopreservation-challenges-in-mesenchymal-stem-cell-culture/?srsltid=AfmBOoqRMfrepv9UVUUWDy4hVnBi-NOaUG-rzckBLGE5rbyiA22UKaAd]
- 118. Validating the cryopreservation of tissue engineered ... [https://www.academia.edu/76200728/Validating_the_cryopreservation_of_tissue_engineered_constructs_within_cryobags]
- 119. The tiny giants of regeneration: MSC-derived extracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12310703/]
- 120. Functionalized mesenchymal stem cells for enhanced bone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577075/]
- 121. Mesenchymal stem cell therapy for type 2 diabetes [https://pubmed.ncbi.nlm.nih.gov/41117868/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1nq_IKqRo1lpVmZPLMBh8BLnGSlMeRq2hML5UR5CAPbykwTYAQ&fc=None&ff=20251024013741&v=2.18.0.post22+67771e2]
- 122. Adipose-derived mesenchymal stromal/stem cells in type 1 ... [https://www.nature.com/articles/s42003-025-08244-z]
- 123. Dimethyl sulfoxide in cryopreserved mesenchymal stromal ... [https://pubmed.ncbi.nlm.nih.gov/40826088/]