Decoding Cryo-Injury: Mechanisms, Prevention, and Functional Impact on Mesenchymal Stem Cell Cryopreservation
This comprehensive review elucidates the multifaceted mechanisms of cryo-injury that threaten Mesenchymal Stem Cell (MSC) viability and function during cryopreservation, a critical process for clinical-grade cell therapy.
Decoding Cryo-Injury: Mechanisms, Prevention, and Functional Impact on Mesenchymal Stem Cell Cryopreservation
Abstract
This comprehensive review elucidates the multifaceted mechanisms of cryo-injury that threaten Mesenchymal Stem Cell (MSC) viability and function during cryopreservation, a critical process for clinical-grade cell therapy. We systematically detail the three primary damage pathways—osmotic stress, intracellular ice crystal formation, and oxidative damage—and their cellular consequences. The article further compares established and emerging cryopreservation methodologies, including slow freezing and vitrification, while evaluating the roles and toxicities of various cryoprotectants like DMSO. It provides actionable strategies for protocol optimization to minimize cryodamage and synthesizes current evidence on the post-thaw functional retention of MSCs, including immunomodulatory capacity and in vivo therapeutic efficacy. This resource is tailored for researchers, scientists, and drug development professionals engaged in biobanking and the development of off-the-shelf MSC-based therapeutics.
The Cellular Battle Against Cold: Unraveling the Core Mechanisms of Cryo-Injury in MSCs
The Critical Role of Cryopreservation in MSC-Based Therapies
Mesenchymal stem/stromal cells (MSCs) have emerged as a highly promising strategy in regenerative medicine due to their self-renewal, pluripotency, and immunomodulatory properties [1]. These nonhematopoietic, multipotent stem cells can differentiate into various mesodermal lineages and modulate the immune system, making them attractive candidates for treating a broad spectrum of human diseases [1]. The therapeutic application of MSCs necessitates efficient long-term storage strategies, as the cells must often be transported from manufacturing facilities to clinical sites while maintaining viability and functionality.
Cryopreservation represents an indispensable method for the preservation and pooling of MSCs to obtain the cell counts required for clinical applications [2]. Without cryopreservation, cells require continuous passage in culture, which can lead to detrimental changes including altered DNA methylation levels, epigenetic modifications such as telomere shortening, and random loss of genomic regions [3]. Cryopreservation in liquid nitrogen at -196°C enables long-term storage while maintaining cellular properties and genetic characteristics, providing a vital cellular resource for therapeutic research and application [3]. This approach offers significant practical advantages, including sufficient time for rigorous quality control testing, off-the-shelf availability, consistent dosing from large-scale cell cultures, and extended geographic reach of viable cell therapies [4].
Fundamental Cryoinjury Mechanisms in MSCs
Physical and Biochemical Damage Pathways
The process of cryopreservation introduces multiple stressors that can compromise MSC viability and function through distinct mechanisms. During freezing, the change of phase from liquid to solid introduces extreme temperature fluctuations as part of the supercooling event, characterized by a rapid rise and fall in temperature [5]. This uncontrolled freezing can lead to cellular damage through several interconnected pathways:
- Intracellular Ice Formation: When cooling occurs too rapidly, intracellular water does not have sufficient time to exit the cell before freezing, resulting in lethal intracellular ice crystals that disrupt cellular structures [5].
- Osmotic Imbalance: As extracellular ice forms, solutes become concentrated in the remaining liquid, creating hypertonic conditions that draw water out of cells and cause excessive cell shrinkage and membrane damage [3].
- Cryoprotectant Toxicity: While essential for protection, cryoprotective agents (CPAs) like dimethyl sulfoxide (DMSO) can exert toxic effects on cells, particularly at suboptimal concentrations or exposure durations [3] [4].
- Oxidative Stress: The thawing process can generate reactive oxygen species (ROS), leading to oxidative damage as a consequence of osmotic imbalances during rehydration [5].
Cell Cycle-Dependent Cryosensitivity
Recent research has identified a fundamental cryoinjury mechanism related to cell cycle status. S phase MSCs demonstrate exquisite sensitivity to cryoinjury, exhibiting heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function [6]. This vulnerability stems from double-stranded breaks in labile replicating DNA that form during the cryopreservation and thawing processes. This discovery reveals that cryoinjury is not uniformly distributed across cell populations but disproportionately affects actively dividing cells, potentially selecting for specific subpopulations and altering the therapeutic characteristics of cryopreserved MSC products [6].
Structural and Functional Compromise
The cumulative effect of these damage pathways extends beyond immediate cell death to include subtler functional impairments. Cryopreservation can alter MSC membrane integrity, receptor presentation, mitochondrial function, and secretory profiles [5]. These changes may diminish the immunomodulatory capacity, differentiation potential, and overall therapeutic efficacy of MSCs, even in cells that remain viable post-thaw [7]. The susceptibility to cryoinjury appears to vary between MSC sources, with bone marrow-derived, adipose-derived, and umbilical cord-derived MSCs potentially exhibiting different resilience profiles [1].
Current Cryopreservation Methodologies
Slow Freezing: The Conventional Approach
Slow freezing remains the recommended technique for clinical and laboratory MSC cryopreservation due to its operational simplicity and minimal contamination risk [3]. The methodology involves several carefully controlled stages:
- CPA Addition: MSCs are mixed with CPAs, typically permeating agents like DMSO combined with non-permeating agents such as sucrose or trehalose [3].
- Controlled Cooling: Cells are cooled at precisely controlled rates, usually within -1°C to -3°C/min, using programmable freezing equipment [3] [5].
- Gradual Temperature Reduction: Samples are initially cooled to -20°C to -80°C before final transfer to long-term storage in liquid nitrogen at -196°C [3].
The slow cooling rate allows gradual cellular dehydration, reducing intracellular ice formation by permitting water to exit cells before freezing [3]. Approximately 70-80% of cells survive when employing this gradual freezing procedure [3]. The success of slow freezing depends heavily on optimizing cooling rates and CPA formulations to balance dehydration with ice crystal damage.
Vitrification: An Alternative Strategy
Vitrification offers an alternative approach by using high concentrations of cryoprotectants and ultra-rapid cooling rates to achieve a glassy, ice crystal-free state [3]. This method employs two distinct strategies:
- Equilibrium Vitrification: Controls CPA concentration and penetration time to allow full cellular dehydration and osmotic equilibrium before freezing [3].
- Non-Equilibrium Vitrification: Uses high CPA concentrations with immediate liquid nitrogen immersion to achieve vitrification almost instantaneously [3].
While vitrification eliminates ice crystal formation, it introduces risks of cryoprotectant-induced chemical toxicity and osmotic shock due to the high CPA concentrations required [3]. The technique also faces challenges with devitrification (ice nucleation upon temperature fluctuations) and requires optimization for larger volume samples [8].
Table 1: Comparison of Primary MSC Cryopreservation Methods
| Parameter | Slow Freezing | Vitrification |
|---|---|---|
| Cooling Rate | -1°C to -3°C/min | Ultra-rapid (>100°C/min) |
| CPA Concentration | Low (5-10%) | High (40-60%) |
| Ice Formation | Extracellular ice crystals | Glassy, ice-free state |
| Primary Damage Mechanisms | Intracellular ice formation, solute effects | CPA toxicity, osmotic shock |
| Typical Post-Thaw Viability | 70-80% [3] | Variable (technology-dependent) |
| Technical Complexity | Moderate | High |
| Suitable Sample Volumes | Wide range | Typically small volumes |
| Current Clinical Adoption | Widespread [3] | Emerging [8] |
Thawing and Post-Thaw Processing
The thawing process represents a critical phase where additional damage can occur. Conventional procedure involves rapidly warming cryopreserved samples in a 37°C water bath until all ice crystals are dissolved [3]. To enhance safety during cell thawing, drying heating equipment may be preferable to water baths due to potential microbial contamination risks [3]. Following thawing, centrifugation is typically required to remove CPAs, particularly toxic agents like DMSO [3]. This post-thaw washing process presents challenges, as it can result in significant cell loss through damage and requires additional manipulation that may affect product functionality and introduce variability [4].
Advanced Strategies for Cryoinjury Mitigation
Cell Cycle Synchronization
A groundbreaking approach to cryoinjury mitigation involves cell cycle synchronization prior to freezing. Research has demonstrated that blocking cell cycle progression at G0/G1 through growth factor deprivation (serum starvation) dramatically reduces post-thaw dysfunction of MSCs by preventing apoptosis induced by double-stranded breaks in replicating DNA [6]. This strategy specifically protects the vulnerable S-phase population, preserving viability, clonal growth, and T-cell suppression function at pre-cryopreservation levels [6]. The effectiveness of this intervention underscores the importance of the cell cycle-dependent cryosensitivity mechanism and provides a targeted method for enhancing post-thaw recovery of therapeutic MSCs.
Ice Nucleation Control
Recent technological advances have addressed the uncontrolled stochastic nature of ice nucleation during freezing. The use of medical-grade ice nucleation inducers (INIs) can significantly increase the mean nucleation temperature from a range of -9.7°C to -16.5°C up to -5.9°C to -9.4°C, reducing the maximum cooling rate from -2.64±0.67°C/min to -2.16±0.05°C/min [5]. This controlled nucleation approach decreases the chaotic effects during freezing, creating a more stable manufacturing process and improving post-thaw recovery of dental pulp MSCs, particularly when nucleation occurs around -10°C [5]. For larger volume cell storage, combining INIs with fast thawing creates the most stable process, while for adhered cells, INIs with slow thawing enable greatest metabolic activity post-thaw [5].
Cryoprotectant Formulation Advances
Innovative approaches to cryoprotectant composition aim to balance protection with reduced toxicity:
- DMSO Optimization: While DMSO remains the preferred cryoprotectant for MSC cryopreservation, strategies to minimize its concentration without compromising efficacy are actively investigated [4].
- Combination Formulations: Mixing permeating agents like DMSO with non-permeating agents such as sucrose or trehalose provides synergistic protection by mitigating osmotic stress [3].
- DMSO-Free Alternatives: Research into alternative cryoprotectants continues, though none have yet demonstrated suitability for clinical application [4].
Table 2: Cryoprotectant Agents and Their Properties in MSC Cryopreservation
| Cryoprotectant | Type | Mechanism of Action | Advantages | Disadvantages |
|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating | Lowers freezing point, improves membrane permeability to water [3] | Effective, widely used, clinical experience [4] | Concentration-dependent toxicity, allergic reactions [3] [4] |
| Glycerol | Permeating | Similar to DMSO | Lower cell toxicity [3] | Inferior cryopreservation effect [3] |
| Sucrose | Non-permeating | Creates hypertonic extracellular environment, promotes dehydration [3] | Reduces required DMSO concentration, stabilizes membranes | Limited effectiveness alone |
| Trehalose | Non-permeating | Stabilizes membranes and proteins, forms glassy state [3] | Protects membrane integrity, can be used intracellularly with special methods | Poor cellular uptake |
Functional Consequences and Assessment of Cryopreserved MSCs
In Vitro Functional Assays
Comprehensive assessment of cryopreserved MSCs requires evaluation beyond simple viability metrics. Standardized functional assays include:
- Clonogenic Potential: Colony-forming unit-fibroblast (CFU-F) assays measure the proliferative capacity and self-renewal capability of MSC populations after thawing [6] [9].
- Multilineage Differentiation: Evaluation of osteogenic, chondrogenic, and adipogenic differentiation potential confirms maintained stem cell functionality [1] [9].
- Immunomodulatory Capacity: PBMC suppression assays and IDO activity measurements determine whether critical therapeutic properties remain intact [7].
- Metabolic Activity: Assessment of mitochondrial function and metabolic status provides insight into cellular health beyond membrane integrity [5].
Recent studies demonstrate that MSC proliferation and multilineage differentiation can be preserved after freezing, with comparable performance between fresh and cryopreserved cells in clonogenic and differentiation assays [9].
In Vivo Therapeutic Efficacy
The ultimate validation of cryopreservation efficacy comes from in vivo models that recapitulate intended clinical applications. Research in various disease models has yielded context-dependent results:
- Positive Outcomes: Cryopreserved MSCs have demonstrated effectiveness in treating disease models of colitis, allergic airway inflammation, and ischemia/reperfusion injury to the eye [7].
- Variable Performance: Cryopreserved MSCs failed to induce a chondrogenic response in a mouse-based chondrocyte-responsive bioassay, suggesting potential limitations for orthopedic applications [7].
- Clinical Observations: In human studies, cryopreserved MSCs have elicited positive responses in clinical trials for critical limb ischemia, while retrospective analysis of GvHD patients suggests fresh MSCs may be more efficacious [7].
Notably, in an osteoarthritis rat model, both fresh and frozen bone marrow aspirate concentrate (BMAC) equally improved histological cartilage scores compared with PBS control, suggesting that the freezing process does not necessarily negate therapeutic potential for cartilage repair [9].
Research Reagent Solutions for MSC Cryopreservation Studies
Table 3: Essential Research Reagents and Materials for MSC Cryopreservation Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| DMSO (Cell Culture Grade) | Primary cryoprotectant | Typically used at 5-10% concentration; requires careful handling and dilution [3] [4] |
| Programmable Controlled-Rate Freezer | Precise control of cooling rates | Enables standardized -1°C/min cooling protocol; reduces batch-to-batch variability [3] [5] |
| Ice Nucleation Device (IND) | Controls ice formation temperature | Increases nucleation temperature to -5°C to -9°C; reduces cooling rate instability [5] |
| Serum-Free Freezing Media | DMSO-free cryopreservation | Alternative strategy to avoid DMSO toxicity; formulation optimization required [4] |
| Annexin V/Propidium Iodide | Viability and apoptosis assessment | Distinguishes live, early apoptotic, and necrotic populations post-thaw [6] |
| IFN-γ Priming Solution | Pre-licensing for enhanced immunomodulation | 48-hour pretreatment before freezing increases IDO expression post-thaw [7] |
| Collagenase/Trypsin Solutions | Tissue dissociation for MSC isolation | Source-dependent optimization required (bone marrow, adipose, umbilical cord) [1] |
| Ficoll Gradient Medium | Mononuclear cell isolation | Density gradient separation for MSC purification from heterogeneous cell mixtures [9] |
Future Perspectives and Concluding Remarks
The field of MSC cryopreservation continues to evolve with several promising research directions. The emerging understanding of cell cycle-dependent cryosensitivity opens new avenues for targeted interventions, such as specific cell cycle synchronization techniques that could further enhance post-thaw recovery [6]. Advanced technologies like controlled ice nucleation demonstrate how engineering solutions can address fundamental biophysical challenges in cryopreservation [5]. The ongoing development of DMSO-free cryopreservation formulations represents another critical frontier, potentially eliminating concerns about cryoprotectant toxicity while maintaining cellular function [4].
Future advances will likely require integrated approaches that combine biological, chemical, and engineering insights. The optimization of cryopreservation protocols must be context-specific, recognizing that different therapeutic applications may demand distinct functional attributes from MSC products [7]. As our understanding of the precise mechanisms underlying MSC therapeutic efficacy improves, cryopreservation strategies can be increasingly tailored to preserve the specific functions required for particular clinical indications.
In conclusion, cryopreservation remains an essential enabling technology for the clinical translation of MSC-based therapies. While significant challenges persist, recent advances in understanding fundamental cryoinjury mechanisms and developing targeted mitigation strategies provide promising pathways toward more effective and reliable cryopreservation protocols. The continued refinement of these approaches will be crucial for realizing the full therapeutic potential of mesenchymal stem cells in regenerative medicine.
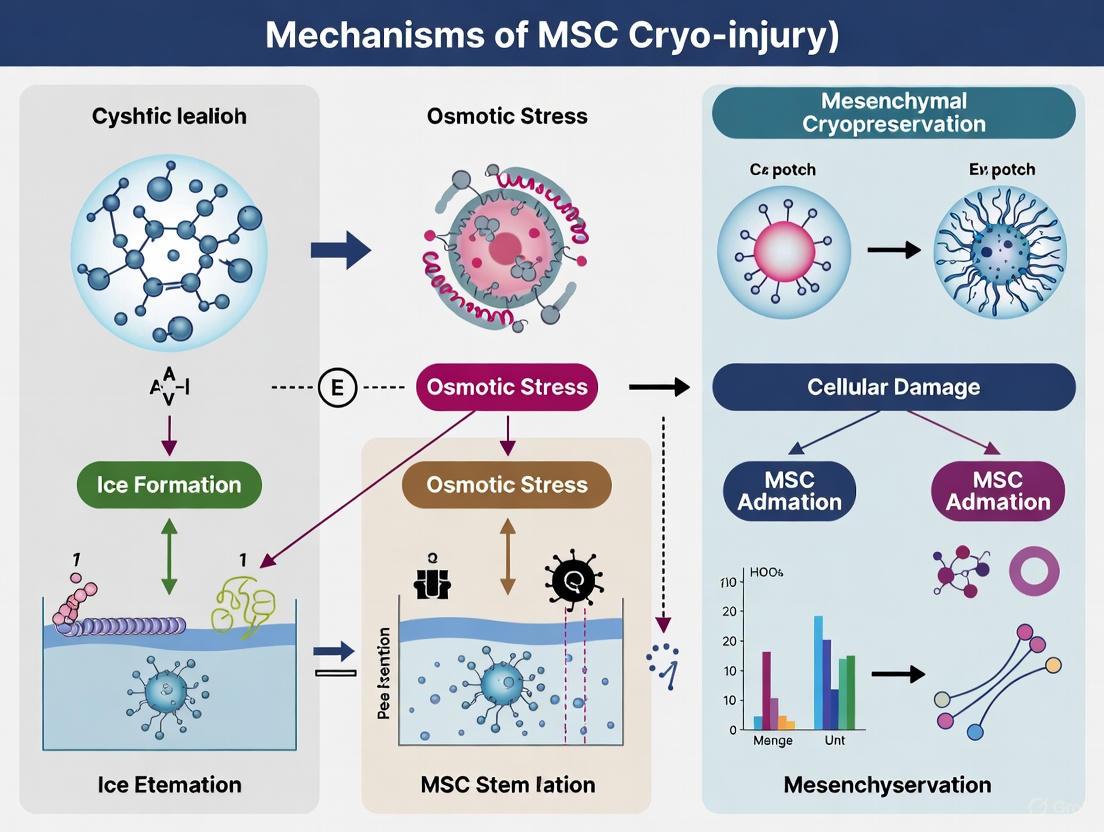
In the context of cryopreservation, osmotic damage refers to the injury inflicted upon cells due to volumetric changes and solute concentration effects that occur during the freezing and thawing processes. For Mesenchymal Stem Cells (MSCs), which serve as crucial therapeutic agents in regenerative medicine, understanding and mitigating this damage is essential for maintaining post-thaw viability and function [10] [11]. When cells are cooled to subzero temperatures, the formation of extracellular ice crystals causes the concentration of solutes in the unfrozen fraction to rise dramatically, creating a hypertonic environment [12]. This imbalance generates an osmotic pressure gradient across the cell membrane, prompting water to exit the cell—a process known as cellular dehydration or shrinkage [13] [14]. The extent of shrinkage is governed by the permeability of the cell membrane to water and the cooling rate. While some dehydration is necessary to avoid the lethal formation of intracellular ice, excessive water loss can lead to a critical reduction in cell volume, damage to the plasma membrane and intracellular structures, and a potentially fatal increase in intracellular solute concentration [12]. This whitepaper provides an in-depth technical analysis of the mechanisms of osmotic injury, summarizes key quantitative data, details relevant experimental methodologies, and discusses emerging strategies to minimize this fundamental cryoinjury in MSCs.
Fundamental Principles of Osmotic Behavior
Physical Chemistry of Water and Solute Transport
The movement of water across cell membranes during cryopreservation is a passive process driven by osmotic gradients. According to the principles of osmosis, water will move from an area of lower solute concentration (higher water potential) to an area of higher solute concentration (lower water potential) across a semi-permeable membrane [14]. During freezing, the formation of extracellular ice effectively removes pure water from the solution, thereby increasing the concentration of all dissolved solutes in the remaining unfrozen fraction. This creates a hypertonic environment outside the cell, establishing an osmotic gradient that draws water out of the cell interior. The rate and extent of this cellular dehydration are determined by the cell's membrane permeability to water and the cooling rate [12]. A slow cooling rate allows sufficient time for water to leave the cell, resulting in progressive dehydration and shrinkage. In contrast, a rapid cooling rate does not provide adequate time for water efflux, leading to the supercooling of the intracellular contents and ultimately, the formation of intracellular ice, which is almost universally lethal to the cell [12] [11].
The Role of Cryoprotective Agents (CPAs)
Cryoprotective Agents (CPAs) are compounds used to protect cells from damage during freezing. They are broadly categorized into two groups based on their ability to cross the cell membrane, and their primary role is to mitigate the harmful effects of increased solute concentration and minimize ice crystal formation [12].
Permeating Agents, such as Dimethyl Sulfoxide (DMSO), glycerol, ethylene glycol (EG), and propylene glycol (PG), are small, typically amphiphilic molecules that can cross the cell membrane [12]. They function by several mechanisms:
- Colligative Action: They depress the freezing point of water and reduce the amount of ice formed at any given temperature. This directly lessens the degree of solute concentration in the unfrozen fraction.
- Reducing Dehydration: By permeating the cell, they increase the intracellular solute concentration. This reduces the osmotic difference between the intra- and extracellular environments, thereby limiting the extent of water efflux and cellular shrinkage [12].
- Promoting Vitrification: At high concentrations and with rapid cooling, they enable the solution to solidify into a glassy, non-crystalline state (vitrification), avoiding ice formation altogether [10].
Non-Permeating Agents, such as sucrose, trehalose, and high molecular weight polymers like polyethylene glycol (PVP), do not enter the cell [12]. They exert their protective effect extracellularly by:
- Inducing Osmotic Dehydration: Prior to freezing, they can draw some water out of the cell, thus reducing the chance of intracellular ice formation.
- Colligative Effects: They contribute to the total solute concentration in the extracellular medium, reducing the amount of ice formed.
- Stabilizing Membranes: Sugars like trehalose are known to interact with phospholipid heads, helping to stabilize the cell membrane in a dry state, a phenomenon known as the "water replacement" hypothesis [12].
Table 1: Common Cryoprotective Agents and Their Properties
| CPA Name | Type | Typical Working Concentration | Mechanism of Action | Reported Toxicity |
|---|---|---|---|---|
| DMSO | Permeating | 10% (v/v) | Depresses freezing point, increases membrane permeability, promotes vitrification [12]. | Cytotoxic at high concentrations/ prolonged exposure; can cause allergic reactions in patients [10]. |
| Glycerol | Permeating | 10-20% (v/v) | Colligative action, reduces osmotic shock [12]. | Lower toxicity than DMSO but generally less effective for many mammalian cells [12]. |
| Ethylene Glycol | Permeating | ~4-6 M (in mixtures) | Rapid permeation, often used in vitrification mixtures [12]. | Toxicity profile similar to DMSO [12]. |
| Trehalose | Non-Permeating | 0.2-0.5 M | Stabilizes membranes, induces protective dehydration, contributes to vitrification [12]. | Very low toxicity; requires specific methods to deliver intracellularly for full efficacy. |
| Sucrose | Non-Permeating | 0.1-0.3 M | Osmotic buffer, used during CPA addition/removal to control cell volume excursions [10]. | Very low toxicity. |
Quantitative Analysis of Osmotic Behavior in MSCs
Experimental studies on human MSCs (hMSCs) have provided critical quantitative data to model their osmotic responses, which is essential for optimizing cryopreservation protocols.
Key Osmotic Parameters
Research on hMSCs isolated from umbilical cord blood has revealed specific biophysical properties. These cells behave as imperfect osmometers, meaning their volume changes do not strictly follow the Boyle-van't Hoff relation predicted by a simple two-parameter model [13]. A significant finding is that during osmotic excursions—such as the shrink-swell process following DMSO addition or the restoration of isotonic conditions—the inactive cell volume fraction appears to increase [13]. This suggests a complex physiological adaptation, potentially involving the activation of mechano-sensitive ion channels, which limits extreme volumetric changes and protects the cell from lysis or severe damage [13].
Table 2: Experimentally Determined Osmotic Parameters for Human MSCs from Umbilical Cord Blood
| Parameter | Symbol | Value at 22°C | Value at 10°C | Value at 4°C | Notes |
|---|---|---|---|---|---|
| Hydraulic Conductivity | ( L_p ) | 0.50 ± 0.08 μm/min/atm | 0.28 ± 0.04 μm/min/atm | 0.16 ± 0.03 μm/min/atm | Measures membrane permeability to water [13]. |
| DMSO Permeability | ( P_{DMSO} ) | 0.47 ± 0.07 x 10⁻³ cm/min | 0.30 ± 0.05 x 10⁻³ cm/min | 0.17 ± 0.03 x 10⁻³ cm/min | Measures membrane permeability to the CPA [13]. |
| Activation Energy ((E_a)) | ( E_a ) | 26.3 ± 4.5 kJ/mol (for (L_p)) | Indicates temperature sensitivity of water transport [13]. | ||
| Inactive Cell Volume | ( V_b ) | Increases during shrink-swell cycles | Suggests adaptive cell response to osmotic stress [13]. |
The data in Table 2 demonstrates that the membrane permeability of hMSCs to both water and DMSO is highly temperature-dependent. As temperature decreases, the permeability drops significantly, which must be accounted for when designing cooling protocols. The low permeability to DMSO relative to water is a key factor necessitating controlled, step-wise addition and removal of CPAs to prevent damaging volume excursions [13] [10].
Experimental Protocol: Measuring Osmotic Parameters
The following methodology, adapted from a study on hMSCs, outlines how to determine the key osmotic parameters listed above [13].
Objective: To determine the hydraulic conductivity ((Lp)), solute permeability ((Ps)), and inactive cell volume ((V_b)) of MSCs.
Materials:
- Cell Source: hMSCs (e.g., from umbilical cord blood, bone marrow).
- Equipment: Impedance-based cell analyzer (e.g., Coulter Counter Multisizer), temperature-controlled bath or stage, data acquisition system.
- Reagents: Isotonic solution (e.g., PBS), hypertonic solution (e.g., PBS with added impermeant solute like NaCl or sucrose), CPA solution (e.g., DMSO in isotonic buffer).
Procedure:
- Cell Preparation: Harvest and suspend MSCs in an isotonic solution at a known concentration. Keep the suspension at the desired experimental temperature (e.g., 4°C, 10°C, 22°C).
- Equilibrium Size Distribution: Measure the cell diameter distribution under isotonic conditions using the impedance analyzer to establish the baseline volume.
- Hypertonic Challenge: Rapidly mix the cell suspension with an equal volume of a pre-cooled hypertonic solution. The final concentration of the impermeant solute should be high enough to induce significant shrinkage (e.g., 1M NaCl).
- Dynamic Sizing: Immediately after mixing, continuously monitor the cell volume (via impedance) over time until a new stable volume is reached. This shrinking phase reflects water efflux.
- Return to Isotonicity (Swelling): After equilibrium is reached in the hypertonic solution, rapidly dilute the suspension with a large volume of pre-cooled isotonic solution. Continuously monitor the subsequent increase in cell volume as water re-enters the cells.
- CPA Addition/Removal: Repeat steps 3-5, but use a solution containing a permeating CPA (e.g., 1M DMSO) instead of an impermeant solute. In this case, the initial shrinkage is followed by a swelling phase as the CPA permeates the cell, bringing water with it. Upon dilution, water rushes in faster than the CPA can leave, causing transient swelling before the cell returns to its original volume.
- Data Fitting: The time-course of volume change is fitted to a mathematical model of mass transport across the membrane (e.g., a two-parameter model or a model incorporating an adaptive inactive volume). The fit of the shrinking curve primarily yields (Lp), while the swelling phase in CPA experiments provides data to calculate (Ps).
This protocol allows researchers to quantify the fundamental osmotic properties of their specific MSC population, which can then be used to inform the development of optimized, cell-specific cryopreservation protocols.
Advanced Research and Emerging Mitigation Strategies
Interplay with Other Cryoinjury Mechanisms
Osmotic damage does not occur in isolation. Recent research has identified a fundamental cryoinjury mechanism in MSCs linked to the cell cycle. Cells in the S phase (DNA replication) are exquisitely sensitive to cryopreservation, demonstrating heightened levels of delayed apoptosis post-thaw [6] [15]. The osmotic and mechanical stresses of freezing and thawing appear to induce double-stranded breaks in the labile, replicating DNA, leading to functional impairment. This underscores that the success of a cryopreservation protocol depends on managing both physical-osmotic stresses and biological vulnerabilities [6].
Another advanced concept is the role of cell-matrix interactions. Studies using microfluidic bioreactors have shown that applying a regulated, low-level shear stress to adherent MSCs before freezing upregulates the formation of focal adhesion points (FPA) [16]. Enhanced FPA expression, measured through proteins like vinculin, is correlated with significantly improved post-thaw cellular survivability. This suggests that reinforcing the cell's mechanical linkage to its substrate can improve its resilience to osmotic and freezing stresses [16].
Strategies to Minimize Osmotic Damage
Based on the current understanding of osmotic principles and MSC biology, several strategies can be employed to mitigate damage:
- Optimized CPA Cocktails: Moving away from single-agent protocols towards mixtures of permeating and non-permeating CPAs. This allows the use of lower, less toxic concentrations of permeating agents like DMSO while maintaining efficacy. For example, combining DMSO with trehalose or sucrose can improve outcomes [12] [16].
- Controlled Rate Freezing: Using a programmable freezer to enforce a slow, controlled cooling rate (typically around -1°C/min) is standard practice for MSCs. This provides adequate time for cellular dehydration without being so slow as to exacerbate solute toxicity or "solution effects" injury [10] [11].
- Cell Cycle Synchronization: Pre-treatment of MSC cultures through growth factor deprivation (serum starvation) to synchronize the population in the G0/G1 phase, which is more resistant to cryoinjury, has been shown to dramatically reduce post-thaw apoptosis and preserve immunomodulatory function [6] [15].
- Stepwise CPA Addition and Removal: Gradually adding and removing CPAs, often with the osmotic support of non-permeating agents like sucrose, prevents the severe volume fluctuations that can cause mechanical damage to the membrane [10].
- Biophysical Priming: Pre-conditioning cells with mild physical stimuli, such as controlled shear stress in a bioreactor to enhance focal adhesions, represents a novel approach to strengthen cells against subsequent cryopreservation stresses [16].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for Investigating Osmotic Damage in MSCs
| Item | Function/Application | Example from Literature |
|---|---|---|
| DMSO | Standard permeating CPA; used to protect against ice formation and reduce osmotic shock [12] [10]. | Used at 10% (v/v) in freezing medium for hMSCs [13] [10]. |
| Trehalose | Non-permeating CPA; stabilizes cell membranes, used in CPA cocktails to reduce toxicity of permeating agents [12] [16]. | Combined with DMSO for enhanced cryopreservation of MSCs in microfluidic devices [16]. |
| Fetal Bovine Serum (FBS) | Component of freezing medium; provides proteins and other macromolecules that can confer membrane-stabilizing and protective effects [13]. | Used at 20-90% in freezing media for hMSCs [13]. |
| Coulter Counter / Impedance Analyzer | To measure cell volume changes in real-time under dynamic osmotic conditions [13]. | Used to track volume excursions of hMSCs exposed to hypertonic solutions and CPAs [13]. |
| Programmable Controlled-Rate Freezer | To apply a consistent, optimized cooling profile (e.g., -1°C/min) crucial for controlled dehydration [10] [11]. | Essential for the standard slow-freezing protocol for MSCs [10]. |
| Microfluidic Bioreactor | To apply controlled shear stress for studying and priming cell-substrate interactions prior to cryopreservation [16]. | Used to demonstrate that shear stress (0.002-0.004 μbar) upregulates FPAs and improves hMSC recovery [16]. |
| Flow Cytometry Antibodies | For immunophenotypic characterization of MSCs pre- and post-cryopreservation to ensure identity is maintained (e.g., CD105, CD73, CD90 positive; CD45, CD34 negative) [13] [16]. | Confirmed MSC marker expression was retained after cryopreservation under shear stress [16]. |
Visualizing Osmotic Damage and Mitigation Pathways
The following diagram illustrates the key mechanisms of osmotic damage during cryopreservation and the primary strategies proposed to mitigate them.
Osmotic damage resulting from cellular dehydration and shrinkage constitutes a primary mechanism of cryoinjury in MSC cryopreservation. The complex osmotic behavior of MSCs, characterized by temperature-dependent membrane permeabilities and adaptive volumetric responses, necessitates a refined approach beyond simplistic models. The integration of quantitative biophysical data—such as hydraulic conductivity and solute permeability—into the design of freezing protocols is critical for success. Furthermore, emerging strategies that address biological vulnerabilities, such as cell cycle synchronization, and those that enhance cellular resilience through biophysical priming, represent the next frontier in cryopreservation research. By synthesizing insights from physical chemistry, cell biology, and engineering, researchers can develop advanced, high-fidelity cryopreservation methods that ensure the delivery of functionally potent MSCs for clinical therapies.
Within the field of Mesenchymal Stem Cell (MSC) cryopreservation research, cryoinjury presents a significant barrier to clinical translation. The formation of ice crystals during freezing and thawing processes is a primary mechanism of mechanical damage that can compromise cell viability and function. Mechanical cryoinjury manifests through two principal physical phenomena: intracellular ice formation (IIF), which is frequently lethal to cells, and the damaging growth of extracellular ice, which imposes mechanical and osmotic stress [17]. Understanding the mechanisms underlying these events is crucial for developing optimized cryopreservation protocols that maintain the therapeutic potency of MSCs post-thaw. This guide provides an in-depth technical examination of ice formation as a mechanism of cryo-injury, framing it within the broader context of ensuring MSC quality for research and clinical applications.
Core Mechanisms of Ice Crystal Damage
The formation of ice crystals during cooling and warming is a complex process governed by thermodynamic and kinetic factors. The damage incurred is largely determined by the cooling rate, which dictates the location and morphology of the ice that forms.
Intracellular Ice Formation (IIF)
IIF is widely regarded as a primary cause of lethal cryoinjury during rapid cooling [18] [19]. When the cooling rate is too rapid, the cell does not have sufficient time to dehydrate in response to the increasing solute concentration in the extracellular space. Consequently, the supercooled intracellular water reaches a critical point where it nucleates, forming ice crystals within the cytoplasm. These crystals can physically disrupt organelles, the cytoskeleton, and the plasma membrane, leading to immediate cell death [17] [20].
The mechanism of IIF initiation is an area of active investigation. While it was historically hypothesized that ice might propagate through membrane pores, recent studies suggest a more complex interplay. Research on cell pairs indicates that the presence of intercellular junction proteins (e.g., for gap, adherens, and tight junctions) may surprisingly result in lower intracellular ice formation temperatures compared to cells lacking these junctions [18]. This counterintuitive finding suggests that the architecture of the cell-cell interface modulates the penetration of extracellular ice into the paracellular space, which in turn influences the probability of intracellular ice nucleation. An alternative hypothesis proposes that the plasma membrane may be damaged by a critical gradient in osmotic pressure across the membrane, thereby nucleating the cytoplasm [19].
Extracellular Ice Formation
During slow cooling, ice typically forms first in the extracellular solution. This initiates an osmotic gradient; as pure water freezes out, the concentration of solutes in the unfrozen extracellular fluid increases. Driven by this osmotic difference, water moves out of the cell, leading to progressive cellular dehydration and volumetric shrinkage [3] [21]. While this process avoids the lethal danger of IIF, it subjects the cell to other forms of injury, collectively known as "solution effects" injury.
The mechanical and structural consequences of extracellular ice are significant:
- Mechanical Stress: The growth of extracellular ice crystals can mechanically compress and deform cells, potentially rupturing the plasma membrane or delicate cellular projections [17].
- RER Ultrastructural Damage: In slow-frozen cells with inadequate cryoprotection, extensive conversion of rough endoplasmic reticulum (RER) into sphere-like vesicles has been observed after thawing, a hallmark of solution-effects injury [20].
- Osmotic Imbalance: The profound cellular dehydration concentrates intracellular solutes, potentially disrupting protein and membrane structure and leading to osmotic shock during the subsequent thawing process [3] [17].
Table 1: Characteristics of Ice Formation and Associated Injury Mechanisms
| Parameter | Intracellular Ice Formation (IIF) | Extracellular Ice Formation |
|---|---|---|
| Primary Cooling Condition | Rapid cooling | Slow cooling |
| Key Trigger | Insufficient time for cellular dehydration | Extracellular ice nucleation and solute concentration |
| Primary Damage Mechanism | Physical disruption of organelles and membranes by internal crystals | Cellular dehydration (solute effects) and mechanical stress from external crystals |
| Typical Cell Survival | Low (often lethal) [19] | Higher (70-80% with optimized protocols) [3] |
| Role of Cryoprotectants (CPAs) | Suppress IIF by increasing viscosity and reducing nucleation probability | Reduce "solution effects" by colligatively lowering the salt concentration [21] |
Quantitative Data on Cryoinjury and Cryoprotection
A critical step in mitigating cryoinjury is the use of Cryoprotective Agents (CPAs). These compounds function through distinct mechanisms to protect against both intracellular and extracellular ice damage. The choice and concentration of CPA are critical and involve a trade-off between protective efficacy and intrinsic toxicity.
Table 2: Comparison of Common Cryoprotective Agents (CPAs) and Their Properties
| Cryoprotectant | Type | Common Concentration | Mechanism of Action | Remarks on Toxicity and Efficacy |
|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating | 5-10% (v/v) [17] | Penetrates cell, reduces ice crystal formation, colligatively lowers freezing point | Highly effective but cytotoxic; can trigger allergic reactions in patients [3] [17] |
| Glycerol | Permeating | 10-20% (v/v) | Similar to DMSO | Lower cell toxicity but inferior cryopreservation effect compared to DMSO [3] |
| Ethylene Glycol (EG) | Permeating | ~10% (v/v) | Similar to DMSO | Cell toxicity lower than DMSO, similar cryopreservation effect [3] |
| Propylene Glycol (PG) | Permeating | ~10% (v/v) | Similar to DMSO | Cell toxicity lower than DMSO, similar cryopreservation effect to EG [3] |
| Sucrose/Trehalose | Non-Permeating | 0.1-0.5 M | Induces protective dehydration, stabilizes membranes, increases solution viscosity | Low toxicity; often used in combination with permeating CPAs to reduce their required concentration [3] |
Experimental Protocols for Studying Ice Formation
Advancing the understanding of ice formation and its damaging effects relies on sophisticated experimental methodologies. The following are key protocols used in the field.
High-Speed Video Cryomicroscopy of Intracellular Ice
This technique allows for the direct observation of the kinetics and spatial propagation of ice formation within cells.
- Sample Preparation: Culture cells of interest (e.g., mouse insulinoma MIN6 cells, hepatoma HepG2 cells) on a specialized cryomicroscopy stage. For cell-cell interaction studies, use micropatterned surfaces to create controlled two-cell pairs [18].
- Junctional Modulation: To investigate the role of cell-cell junctions, treat cell pairs with specific inhibitors (e.g., 18β-glycyrrhetinic acid for gap junctions) or use genetic knock-down strains [18].
- Freezing and Data Acquisition: Place the stage on a controlled cooling apparatus. Cool samples at a defined rapid rate (e.g., 130°C/min) while recording with a high-speed video camera (≥1000 frames per second) [18] [19].
- Data Analysis: Analyze video recordings to determine the temperature of ice initiation, the sequence of freezing between cells in a pair, and the physical location (e.g., paracellular space) from which intracellular ice nucleates. Data can be further analyzed using probabilistic models like Markov chains to deduce propagation mechanisms [18].
Slow Freezing and Post-Thaw Viability Assessment for MSCs
This protocol outlines a standard slow-freezing method and subsequent evaluation of cryodamage in MSC populations.
- Cell Preparation and CPA Addition: Harvest and resuspend MSCs (e.g., from bone marrow or adipose tissue) in a suitable base medium. Gently add pre-chilled freezing medium containing a permeating CPA like 10% DMSO, potentially combined with a non-permeating agent like sucrose. Common practice is to use a controlled-rate freezer [3] [17].
- Controlled-Rate Freezing: Cool the cell suspension from room temperature to 4°C, then further to -80°C at a controlled rate of approximately -1°C/min to -3°C/min. Finally, transfer the samples to liquid nitrogen (-196°C) for long-term storage [3].
- Thawing and CPA Removal: Rapidly thaw the vial in a 37°C water bath until the last ice crystal disappears. Dilute the cell suspension step-wise with pre-warmed culture medium to reduce CPA concentration gradually and prevent osmotic shock. Centrifuge to remove the CPA-containing supernatant and resuspend the cell pellet in fresh medium [3] [17].
- Post-Thaw Analysis:
- Viability: Assess immediately using dye exclusion tests (e.g., Trypan Blue).
- Functionality: Perform clonogenic assays (CFU-f) and multi-lineage differentiation assays (osteogenic, adipogenic, chondrogenic) days after thawing to determine functional retention [9].
- Cryoinjury Markers: Evaluate delayed-onset apoptosis and DNA double-stranded breaks (e.g., via γH2AX staining) days after thawing, as these are key indicators of cryoinjury, particularly in sensitive S-phase cells [6].
Mitigation Strategies and the Scientist's Toolkit
The overarching goal of cryopreservation research is to develop strategies that minimize the mechanical and related damage described above. The following diagram synthesizes the key factors and relationships involved in the decision-making process for an MSC cryopreservation protocol, highlighting the balance between different injury mechanisms.
Effective mitigation of cryoinjury requires a suite of specialized reagents and tools. The table below details essential items for a research laboratory focused on MSC cryopreservation.
Table 3: Research Reagent Solutions for MSC Cryopreservation Studies
| Tool/Reagent | Function/Explanation | Example Application |
|---|---|---|
| Controlled-Rate Freezer | Precisely controls cooling rate (e.g., -1°C/min), which is critical for reproducible slow-freezing protocols and avoiding IIF. | Standardized slow freezing of MSC aliquots for biobanking [3]. |
| Permeating CPAs (e.g., DMSO) | Penetrate the cell, reduce ice crystal formation colligatively, and promote vitrification. | Primary cryoprotectant in freezing medium (5-10% final concentration) [3] [17]. |
| Non-Permeating CPAs (e.g., Sucrose) | Do not enter the cell; induce protective dehydration and stabilize membranes osmotically. | Used in combination with DMSO to reduce its required concentration and toxicity [3]. |
| Serum-Free Freezing Media | Chemically defined media formulations designed to support cell stability during freeze-thaw, often with reduced DMSO. | Clinical-grade MSC cryopreservation to minimize variability and animal-derived components. |
| High-Speed Video Cryomicroscope | Allows direct visualization of ice formation dynamics in real-time at the cellular level. | Investigating the kinetics of IIF propagation between cells in a monolayer [18] [19]. |
| Cell Cycle Synchronization Agents | Agents like serum starvation halt cells in G0/G1 phase, making them less susceptible to cryo-induced DNA damage. | Pre-treatment of MSC cultures before freezing to enhance post-thaw viability and function [6]. |
Mechanical damage from intracellular and extracellular ice formation represents a fundamental challenge in MSC cryopreservation. The cooling rate directly dictates the dominant injury pathway, creating a critical trade-off between the lethal internal ice crystals of rapid cooling and the dehydrating solute effects of slow cooling. Contemporary research, leveraging tools like high-speed cryomicroscopy, continues to refine our understanding of these physical mechanisms, revealing unexpected complexities such as the role of cell-cell junctions in IIF. The ultimate goal is the rational design of cryopreservation protocols—through optimized cooling rates, CPA combinations, and novel cell pre-conditioning strategies—that can effectively navigate these hazards. Success in this endeavor is paramount to delivering fully functional MSCs from the freezer to the patient, thereby unlocking the full clinical potential of regenerative medicine.
Reactive oxygen species (ROS)-induced oxidative stress is a major mediator of cryo-injury in mesenchymal stem cell (MSC) cryopreservation. During freezing and thawing, supraphysiological ROS levels trigger lipid peroxidation, DNA damage, and apoptotic signaling, compromising cell viability and function [22] [23]. This whitepaper synthesizes mechanisms of ROS generation, antioxidant defenses, and experimental strategies to mitigate oxidative stress in MSC cryopreservation, providing a technical framework for researchers and drug development professionals.
Mechanisms of ROS Generation and Oxidative Stress
- Mitochondrial Electron Transport Chain: Electron leakage during freeze-thaw cycles generates superoxide anions (O₂•⁻) [22].
- Membrane-Bound Enzymes: NADPH oxidase (NOX) and xanthine oxidase (XO) produce O₂•⁻ and H₂O₂ [23].
- Exogenous Triggers: Cryoprotectant toxicity (e.g., DMSO), ambient oxygen, and metal ions (e.g., Fe²⁺) exacerbate ROS via Fenton/Haber-Weiss reactions [23] [24].
ROS-Induced Damage Pathways
- Lipid Peroxidation: ROS attack polyunsaturated fatty acids in plasma membranes, releasing malondialdehyde (MDA) and disrupting membrane integrity [22] [24].
- DNA Fragmentation: Hydroxyl radicals (•OH) cause single/double-strand breaks, impairing genomic stability [22].
- Protein Oxidation: Sulfhydryl group oxidation and carbonylation alter enzymatic activity and signal transduction [23].
Apoptotic Signaling
ROS activate intrinsic apoptosis via mitochondrial permeability transition pore opening, cytochrome c release, and caspase-3 activation. The BAX/BCL-2 ratio serves as a key apoptotic indicator [24].
Diagram: ROS-Mediated Apoptotic Signaling in Cryopreserved MSCs
Quantitative Data on ROS and Cryo-Injury
Table 1: Biomarkers of Oxidative Stress in Cryopreserved Cells
| Biomarker | Change Post-Cryopreservation | Detection Method | Functional Impact |
|---|---|---|---|
| Lipid Peroxidation (MDA) | ↑ 2–3 fold [24] | Thiobarbituric acid assay | Membrane integrity loss |
| DNA Fragmentation | ↑ 40–60% [22] | TUNEL/Comet assay | Impaired genomic integrity |
| Mitochondrial Membrane Potential | ↓ 50–70% [23] | JC-1 staining | Reduced ATP production |
| BAX/BCL-2 Ratio | ↑ 3–4 fold [24] | qPCR/Western blot | Apoptosis activation |
Table 2: Efficacy of Antioxidants in Mitigating Cryo-Injury
| Antioxidant | Concentration | Cell Type | Outcome | Mechanism |
|---|---|---|---|---|
| Melatonin [25] | 0.1–1 mM | Rat Ovarian Tissue | ↑ Viability (70–85%) | Activates Nrf2 pathway |
| Ascorbic Acid [23] | 50–100 µM | Spermatozoa | ↓ DNA damage | Direct ROS scavenging |
| MSC-Derived Exosomes [24] | 1.5 µg/mL | Canine Sperm | ↑ Motility (60.3%) | miRNA-mediated protection |
| Catalase [23] | 100 U/mL | Spermatozoa | ↓ H₂O₂ (40–50%) | H₂O₂ decomposition |
Experimental Protocols for ROS Assessment
ROS Detection Workflow
Diagram: Experimental Workflow for ROS Analysis
Detailed Methodology
- Cell Preparation: Culture MSCs to 80% confluence. Dissociate with trypsin-EDTA [24].
- Cryopreservation:
- ROS Staining: Incubate thawed cells with 10 µM H2DCFDA (general ROS) or 5 µM DHE (O₂•⁻) for 30 min [23].
- Oxidative Stress Assays:
- Apoptosis Detection:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for ROS Studies in Cryopreservation
| Reagent | Function | Example Application |
|---|---|---|
| H2DCFDA [23] | Fluorescent ROS detection | General oxidative stress measurement |
| Dihydroethidium (DHE) [23] | Superoxide detection | O₂•⁻ quantification in mitochondria |
| Melatonin [25] | ROS scavenger and Nrf2 activator | Improves viability in ovarian tissues |
| MSC-Derived Exosomes [24] | miRNA-mediated protection | Enhances motility in spermatozoa |
| Catalase [23] | H₂O₂ decomposition | Reduces hydroxyl radical formation |
| N-Acetylcysteine (NAC) | Glutathione precursor | Augments intracellular antioxidant capacity |
Antioxidant Defense Pathways
Diagram: Nrf2-Mediated Antioxidant Response
ROS-induced oxidative stress and apoptosis are pivotal in MSC cryo-injury. Integrating antioxidants (e.g., melatonin, exosomes) and monitoring biomarkers (e.g., MDA, BAX/BCL-2) via standardized protocols can significantly improve post-thaw recovery. Future work should focus on engineering novel cryoprotectants that target ROS signaling pathways specifically.
Cryopreservation represents a pivotal technology in regenerative medicine, enabling the long-term biobanking of mesenchymal stem cells (MSCs) for therapeutic applications. Despite technological advances, the process of freezing and thawing inflicts substantial cryo-injury on fundamental cellular structures, compromising membrane integrity, organelle function, and cytoskeletal architecture. These injuries directly diminish post-thaw viability, functionality, and the therapeutic efficacy of MSCs, presenting a significant barrier to clinical translation [10] [26]. Within the broader context of cryo-injury mechanisms, this technical guide provides an in-depth analysis of how cryopreservation impacts core cellular structures in MSCs. We summarize quantitative data on cryo-injury, detail standardized experimental protocols for its assessment, and visualize key molecular pathways involved, providing researchers and drug development professionals with a comprehensive resource to advance cryopreservation methodologies.
Core Mechanisms of Cryo-Injury to Cellular Structures
The cryopreservation process, encompassing freezing, storage, and thawing, subjects cells to severe physical and chemical stresses. The injury mechanisms can be broadly categorized into direct physical damage and indirect biochemical cascades.
Physical Damage from Ice Crystals and Osmotic Stress
During slow freezing, the extracellular solution freezes first, elevating solute concentration and creating a hypertonic environment. This draws water out of the cell, leading to dehydration and excessive cell shrinkage, which can cause membrane lysis [26]. If cooling is too rapid, intracellular water does not have time to exit and forms intracellular ice crystals (IIF). These sharp crystals mechanically disrupt organelles and the plasma membrane, often leading to immediate cell death [10] [26]. The cycle of solute concentration fluctuations during freezing and thawing imposes severe osmotic stress, testing the mechanical limits of the plasma membrane and contributing to its failure [26].
Biochemical and Metabolic Damage
The post-thaw period is characterized by the activation of detrimental biochemical pathways. Oxidative stress is a major contributor, where the generation of reactive oxygen species (ROS) upon thawing damages lipids, proteins, and DNA [8]. Ischemia-reperfusion injury (IRI) parallels this in transplanted tissues, where revascularization generates a burst of ROS, inducing vascular dysfunction and amplifying inflammatory responses [8]. Furthermore, cryopreservation can dysregulate critical survival pathways, such as the mTOR signaling pathway, leading to aberrant cellular processes like premature activation of primordial follicles in ovarian tissue, a phenomenon that may have parallels in MSC population depletion [8]. These insults can trigger programmed cell death pathways, including apoptosis and the recently identified role of pyroptosis in cryo-injury, as evidenced by elevated levels of caspase-1 and NLRP3 in grafted tissues [8].
Table 1: Summary of Primary Cryo-Injury Mechanisms and Their Cellular Targets
| Mechanism | Description | Primary Cellular Structures Affected |
|---|---|---|
| Intracellular Ice Crystallization | Formation of ice crystals inside the cell during rapid cooling. | Plasma membrane, organelle membranes, cytoskeleton. |
| Solution Effects & Dehydration | Concentration of solutes and cellular shrinkage during slow water efflux. | Plasma membrane (lysis), protein denaturation. |
| Oxidative Stress | Overproduction of Reactive Oxygen Species (ROS) during thawing. | Lipid membranes (peroxidation), mitochondrial DNA, proteins. |
| Ischemia-Reperfusion Injury (IRI) | Burst of ROS and inflammatory response upon restoration of blood flow/oxygen. | Mitochondria, endothelial cells, overall cell viability. |
Quantitative Data on Cryo-Induced Structural Damage
The impact of cryopreservation on MSC integrity has been quantified across numerous studies. The following tables consolidate key findings on survival rates and the specific effects on cellular structures and signaling under different preservation protocols.
Table 2: Quantitative Impact of Cryopreservation on MSC Viability and Structure
| Parameter Assessed | Finding | Notes / Method | Reference |
|---|---|---|---|
| Post-Thaw Viability (Slow Freezing) | Approximately 70–80% cell survival. | Viability depends on CPA type and concentration. | [10] |
| Stromal Cell Viability Loss | Significant loss, with viability < 65% post-slow-freezing. | Highlights sensitivity of supportive stromal cells. | [8] |
| Mitochondrial Dysfunction | Disruption of mitochondrial membrane potential homeostasis. | Leads to activation of apoptotic cascades. | [8] |
| Cytoskeletal & Junction Alterations | Diminished follicular morphological integrity; downregulation of gap junction proteins. | Compromises cell-cell communication and structural cohesion. | [8] |
Table 3: Cryoprotectant Agent (CPA) Toxicity and Efficacy Profile
| Cryoprotectant | Class | Relative Toxicity | Key Characteristics | Reference |
|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating | Moderate to High | Gold standard but can trigger allergic responses; cell toxicity increases with temperature. | [10] [26] |
| Glycerol | Penetrating | Lowest | Lower cell toxicity but results in worst cryopreservation effect. | [10] |
| Ethylene Glycol (EG) | Penetrating | Lower than DMSO | Exhibits similar cell toxicity to Propylene Glycol (PG). | [10] |
| Propylene Glycol (PG) | Penetrating | Lower than DMSO | Similar toxicity to EG, but worst cryopreservation effect among the group. | [10] |
| Sucrose / Trehalose | Non-Penetrating | Low | Often used in combination with penetrating CPAs to mitigate osmotic shock. | [10] [26] |
Experimental Protocols for Assessing Cryo-Injury
Standardized methodologies are crucial for the accurate evaluation of cryo-injury to cellular structures. Below are detailed protocols for key assays.
Protocol: Membrane Integrity Assessment via Live/Dead Staining
Principle: This assay uses fluorescent dyes to distinguish between live cells with intact plasma membranes and dead cells with compromised membranes. Reagents: Phosphate Buffered Saline (PBS), Calcein-AM, Ethidium homodimer-1 (EthD-1), Hoechst 33342 (optional). Procedure:
- Thawing: Rapidly thaw frozen MSC vials in a 37°C water bath until only a small ice crystal remains.
- CPA Removal: Gently transfer the cell suspension to a centrifuge tube containing pre-warmed culture medium. Centrifuge at 300 x g for 5 minutes. Carefully aspirate the supernatant.
- Staining Solution: Resuspend the cell pellet in PBS. Prepare a working solution containing 2 µM Calcein-AM and 4 µM EthD-1 in PBS. Add Hoechst 33342 (1 µg/mL) for nuclear counterstaining if desired.
- Incubation: Incubate the cell suspension in the staining solution for 20-30 minutes at room temperature in the dark.
- Analysis: Wash cells with PBS and analyze immediately under a fluorescence microscope or flow cytometer.
- Calcein-AM (green fluorescence, ~515 nm): Metabolically active live cells.
- EthD-1 (red fluorescence, ~635 nm): Penetrates dead cells with damaged membranes.
- Hoechst 33342 (blue fluorescence): Labels all nuclei.
Protocol: Immunofluorescence Analysis of Cytoskeleton and Organelles
Principle: This protocol uses specific antibodies and phalloidin to visualize the organization of intracellular structures, revealing cryo-induced disassembly. Reagents: Paraformaldehyde (4% in PBS), Triton X-100 (0.1% in PBS), bovine serum albumin (BSA, 1-3% in PBS), primary antibodies (e.g., anti-α-Tubulin), fluorescently-labeled secondary antibodies, Phalloidin (e.g., conjugated to Alexa Fluor 488/568), DAPI. Procedure:
- Cell Seeding: Seed post-thaw MSCs on glass coverslips and culture for 4-24 hours to allow for recovery and attachment.
- Fixation: Wash cells with PBS and fix with 4% PFA for 15 minutes at room temperature.
- Permeabilization & Blocking: Wash fixed cells with PBS. Permeabilize with 0.1% Triton X-100 for 10 minutes. Wash again and incubate with 1-3% BSA in PBS for 30 minutes to block non-specific binding.
- Staining: Incubate with primary antibody (diluted in BSA solution) for 1 hour at room temperature or overnight at 4°C. Wash thoroughly with PBS. Incubate with fluorescent secondary antibody and phalloidin for 1 hour in the dark. Wash again.
- Mounting and Imaging: Mount the coverslip on a glass slide using an antifade mounting medium containing DAPI. Seal the edges and image using a confocal or epifluorescence microscope.
Immunofluorescence Staining Workflow
Protocol: Slow Freezing of MSCs
Principle: Controlled-rate freezing allows gradual cellular dehydration, minimizing lethal intracellular ice formation. Reagents: MSC culture, Trypsin/EDTA, Culture medium with serum, Cryoprotective Medium (e.g., 10% DMSO in FBS), Programmable freezer or -80°C freezer. Procedure:
- Harvesting: Harvest MSCs at 80-90% confluence using trypsin/EDTA. Neutralize with complete medium.
- CPA Addition: Pellet cells by centrifugation, resuspend in cryoprotective medium at a concentration of 0.5-2 x 10^6 cells/mL, and aliquot into cryovials.
- Equilibration: Incubate vials on ice for 15-30 minutes.
- Freezing: Place vials in a controlled-rate freezer. Cool at -1°C/min to -80°C. Alternatively, use a "Mr. Frosty" container filled with isopropanol, placed at -80°C for 24 hours.
- Storage: Transfer vials to liquid nitrogen (-196°C) for long-term storage.
Protocol: Vitrification of MSCs
Principle: Ultra-rapid cooling with high CPA concentrations solidifies the cell solution into a glassy state without ice crystallization. Reagents: Base Medium, Equilibration Solution (e.g., 7.5% DMSO + 7.5% EG), Vitrification Solution (e.g., 15% DMSO + 15% EG + 0.5 M Sucrose), Liquid nitrogen, Cryoloops/straws. Procedure:
- Equilibration: Expose MSCs (in a small droplet or on a cryoloop) to the Equilibration Solution for 10-15 minutes at room temperature.
- Vitrification: Transfer cells to the high-concentration Vitrification Solution. Quickly (within 60-90 seconds) plunge the specimen directly into liquid nitrogen.
- Storage: Store in liquid nitrogen under stable conditions to prevent devitrification.
Visualization of Key Signaling Pathways in Cryo-Injury
Cryopreservation-induced stress activates and disrupts several key intracellular signaling pathways, leading to cell death or dysfunction. The diagram below illustrates the central role of the PI3K/AKT/mTOR and oxidative stress pathways in aberrant cellular activation and apoptosis.
Signaling Pathways in Cryo-Injury
The Scientist's Toolkit: Essential Research Reagents
A curated list of critical reagents for investigating cryo-injury to cellular structures is provided below.
Table 4: Key Research Reagent Solutions for Cryo-Injury Studies
| Reagent / Kit | Function / Application | Key Characteristics |
|---|---|---|
| Calcein-AM / EthD-1 Assay | Simultaneous fluorescent staining of live and dead cells. | Assesses plasma membrane integrity; standard for post-thaw viability. |
| DAPI or Hoechst 33342 | Nuclear counterstain. | Labels all nuclei; essential for cell counting and viability normalization. |
| Phalloidin (Fluorescent Conjugates) | High-affinity staining of F-actin. | Visualizes cytoskeletal architecture and stress-induced disassembly. |
| Anti-Tubulin Antibodies | Immunostaining of microtubules. | Evaluates integrity of the microtubule network and centrosomes. |
| MitoTracker Probes | Staining of functional mitochondria. | Assesses mitochondrial mass, membrane potential, and localization. |
| ROS Detection Kits (e.g., H2DCFDA) | Measurement of intracellular reactive oxygen species. | Quantifies oxidative stress levels during thawing and recovery. |
| Annexin V / PI Apoptosis Kit | Flow cytometry detection of apoptotic and necrotic cells. | Distinguishes between early/late apoptosis and necrosis post-thaw. |
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant. | The most common CPA; requires careful handling and removal due to toxicity. |
| Sucrose / Trehalose | Non-penetrating cryoprotectants. | Provides osmotic support; mitigates osmotic shock; used with penetrating CPAs. |
| Caspase-3/7 Activity Assay | Fluorometric or luminescent detection of caspase activity. | Quantifies activation of executioner caspases in apoptosis pathway. |
From Theory to Practice: Cryopreservation Techniques and Cryoprotectant Strategies for MSCs
Slow freezing, or controlled-rate freezing, represents a foundational methodology for the long-term cryopreservation of mesenchymal stem cells (MSCs). This technique centers on precisely managing the cooling rate to mitigate the primary cause of cryoinjury: the formation of intracellular ice crystals. By facilitating controlled cellular dehydration, slow freezing promotes the harmless extrusion of water, thereby minimizing intracellular ice formation (IIF) and its consequent mechanical damage to cellular structures. This technical guide delineates the core principles, detailed protocols, and underlying physiological responses of MSCs to controlled-rate cooling, providing a critical framework for researchers and development professionals aiming to preserve cell viability and function for therapeutic applications.
Cryopreservation is indispensable for creating readily available, quality-controlled banks of MSCs for clinical and research use, overcoming the logistical challenges of continuous in vitro culture and enabling "off-the-shelf" cell therapy products [3] [27]. Among available techniques, slow freezing is the most established method for the cryopreservation of MSC suspensions, prized for its operational simplicity and reduced risk of contamination [3].
The fundamental goal of slow freezing is to navigate the inherent physical dangers of the freezing process, primarily governed by Mazur's "two-factor hypothesis" of cryoinjury [28] [29]. This theory posits that cell survival depends critically on the cooling rate:
- At slow cooling rates, the extracellular solution freezes first. This increases the concentration of solutes outside the cell, creating an osmotic gradient that draws water out of the cell. The cell undergoes progressive dehydration, which is protective as it reduces the amount of water available to form lethal intracellular ice upon further cooling. However, if the cooling is too slow, prolonged exposure to hypertonic conditions can cause "solution damage" or excessive cell shrinkage, damaging the plasma membrane and cytoskeleton [30] [28].
- At rapid cooling rates, water within the cell does not have sufficient time to exit before the intracellular temperature falls below its nucleation point. This results in the formation of intracellular ice crystals, which are almost universally fatal, causing mechanical destruction of organelles and membrane systems [30] [31].
Slow-freezing protocols aim for an optimal cooling rate—typically around -1°C/min to -3°C/min—that balances these two injury mechanisms, favoring sufficient dehydration to avoid intracellular ice while minimizing exposure to solute effects [3] [28].
Physiological Basis: MSC Response to Controlled Cooling
The success of slow freezing hinges on inducing a series of coordinated physiological responses in MSCs to the progressively freezing environment.
The Core Mechanism: Gradual Dehydration
The defining mechanism of slow freezing is gradual cellular dehydration [3]. As the temperature drops in a controlled manner, ice forms preferentially in the extracellular space. Because ice crystals exclude solutes, the unfrozen fraction of the extracellular solution becomes increasingly concentrated with salts and other solutes. This creates a transient osmotic imbalance where the intracellular environment is hypotonic relative to the outside. Water flows out of the cell across the plasma membrane, shrinking the cell and concentrating the intracellular contents. This process effectively depresses the intracellular freezing point and reduces the probability of IIF [3] [31].
The Role of Cryoprotective Agents (CPAs)
Cryoprotective agents are essential components of any freezing medium, and their function is integral to the physiological process. CPAs are classified by their ability to cross the cell membrane:
- Penetrating CPAs (e.g., Dimethyl Sulfoxide - DMSO, glycerol): These are small, neutral molecules that readily diffuse into cells. They function by:
- Reducing the freezing point of both intra- and extracellular solutions.
- Diluting intracellular electrolytes, thereby mitigating "solution damage" from increased salt concentrations.
- Modulating the phase behavior of water, effectively increasing the fraction of unfreezeable water and reducing the amount of ice formed at any given temperature [3] [27].
- Non-Penetrating CPAs (e.g., sucrose, trehalose, hydroxyethyl starch): These large molecules remain outside the cell. They exert a colligative effect on the extracellular solution, further drawing water out of the cell and enhancing dehydration. They also increase the viscosity of the extracellular matrix, which can suppress the growth of ice crystals [3] [27].
The addition and removal of CPAs must be carefully controlled, as rapid osmotic shifts can cause cell swelling or shrinkage, leading to osmotic stress and injury [3].
Molecular and Biochemical Cryoinjury
Beyond immediate physical ice damage, slow freezing imposes other stresses on MSCs:
- Oxidative Stress: The freeze-thaw process can generate excessive reactive oxygen species (ROS), leading to lipid peroxidation, protein oxidation, and DNA damage, which can trigger apoptosis post-thaw [31].
- Cryosensitivity Linked to Cell Cycle: Recent evidence indicates that MSCs in the S-phase (DNA synthesis) of the cell cycle are particularly vulnerable to cryoinjury, demonstrating heightened post-thaw apoptosis and reduced function. This is attributed to double-stranded DNA breaks induced by freezing and thawing in labile, replicating DNA [6].
- Membrane and Cytoskeletal Damage: Osmotic stress and ice crystal formation can disrupt the plasma membrane integrity and the actin cytoskeleton. For adherent cells, this can be especially detrimental to focal adhesion complexes, compromising cell-matrix interactions vital for post-thaw attachment and survival [16].
Standardized Slow-Freezing Protocol for MSCs
The following section provides a detailed, step-by-step experimental methodology for the slow-freezing of mesenchymal stem cell suspensions, representative of protocols used in current research.
Pre-Freezing: Cell Preparation and CPA Addition
- Cell Harvesting: Culture MSCs to the desired passage and confluence. Harvest cells using standard techniques (e.g., trypsin/EDTA for adherent cells), ensuring to neutralize the enzyme activity with serum-containing medium.
- Cell Counting and Centrifugation: Perform a viable cell count and centrifuge the cell suspension to form a pellet. Aspirate and discard the supernatant.
- CPA Medium Resuspension: Resuspend the cell pellet in pre-chilled (4°C) cryopreservation medium at a typical density of (0.5 - 1.0 \times 10^6) cells/mL. A common base formulation is culture medium supplemented with 10% (v/v) DMSO and 10-20% (v/v) fetal bovine serum (FBS) or serum-free alternatives like human platelet lysate. Non-penetrating CPAs like 0.2M sucrose can be added for synergistic protection [3] [32] [27].
- Aliquoting: Quickly aliquot the cell suspension into cryogenic vials (e.g., 1 mL per vial).
- Equilibration: Incubate the filled vials on ice or at 4°C for 15-30 minutes to allow for CPA permeation and temperature equilibration.
Controlled-Rate Freezing Process
- Programmable Cooler: Place the vials into a pre-cooled chamber of a controlled-rate freezer. Initiate the following standard cooling program [3]:
- Hold at 4°C for 10 minutes.
- Cool from 4°C to -20°C at a rate of -1°C/min.
- Cool from -20°C to -80°C at a rate of -3°C/min.
- Hold at -80°C for a minimum of 2 hours (or overnight) to ensure uniform temperature.
- "Mr. Frosty" Alternative: If a programmable freezer is unavailable, use an isopropanol-based freezing container (e.g., "Mr. Frosty"). Place the vials in the container and store it at -80°C for 18-24 hours. This device provides an approximate cooling rate of -1°C/min, which is sufficiently slow for many cell types.
Long-Term Storage and Thawing
- Transfer to LN₂: After the initial freezing step, promptly transfer the vials to the vapor or liquid phase of a liquid nitrogen storage tank (-150°C to -196°C) for long-term preservation [3].
- Rapid Thawing: For thawing, remove the vial from liquid nitrogen and immediately immerse it in a 37°C water bath with gentle agitation until only a small ice crystal remains (typically 2-3 minutes) [3].
- CPA Removal: Decontaminate the vial with 70% ethanol. Gently transfer the cell suspension to a tube containing pre-warmed culture medium (e.g., 10 mL). Centrifuge to pellet the cells and carefully aspirate the supernatant containing the cytotoxic DMSO.
- Post-Thaw Culture: Resuspend the cell pellet in fresh, complete culture medium and plate for subsequent experiments or expansion. Allowing a 24-hour recovery period before analysis is crucial to avoid "false positive" viability readings from early-stage apoptotic cells [33].
Table 1: Key Parameters in a Standard Slow-Freezing Protocol for MSCs
| Protocol Stage | Parameter | Typical Setting | Physiological Rationale |
|---|---|---|---|
| Pre-Freezing | CPA | 10% DMSO + 10-20% FBS | Permeating CPA + protein source for membrane stability |
| Temperature | 4°C | Slows metabolism, reduces CPA toxicity | |
| Equilibration Time | 15-30 min | Permits CPA penetration | |
| Controlled Cooling | Rate (4°C to -20°C) | -1°C/min to -3°C/min | Optimizes cellular dehydration while minimizing IIF |
| Final Cooler Temperature | -80°C | Transition point before long-term storage | |
| Storage | Long-Term Temperature | -150°C to -196°C (LN₂) | Halts all metabolic & biochemical activity |
| Thawing | Method | 37°C Water Bath | Prevents destructive ice recrystallization |
Current Advances and Quantitative Data
Research continues to refine slow-freezing protocols, focusing on novel CPAs and preconditioning strategies to enhance post-thaw recovery.
Emerging Cryoprotectants
Combinations of conventional and novel CPAs show significant promise. For instance, a zwitterion/DMSO solution has demonstrated superior performance in cryopreserving complex systems like cell spheroids and tumor tissues.
Table 2: Efficacy of a Zwitterion/DMSO Solution for Spheroid Cryopreservation [33]
| Cryopreservation Solution (Zwitterion/DMSO/Water) | Relative Cell Recovery (Post-Thaw) | Relative Cell Recovery (After 24h Culture) | Key Finding |
|---|---|---|---|
| ZD-0/10 (0/10/90) | 0.11 | N/A | DMSO alone is ineffective for spheroids |
| ZD-10/0 (10/0/90) | 0.15 | N/A | Zwitterion alone is ineffective for spheroids |
| ZD-5/5 (5/5/90) | 1.32 | 0.73 | "False positive" result; recovery drops after culture |
| ZD-5/15 (5/15/80) | 1.37 | 1.72 | High & sustained recovery, minimal apoptosis |
| ZD-10/15 (10/15/75) | 1.51 | 1.37 | High & sustained recovery, minimal apoptosis |
| Commercial CPA (Control) | 1.00 (Baseline) | 1.00 (Baseline) | Baseline for comparison |
Mitigating Cryoinjury via Cell Cycle Synchronization
A groundbreaking approach to reducing cryoinjury involves synchronizing the cell cycle prior to freezing. Research has identified that S-phase MSCs are exquisitely sensitive to freezing-induced DNA damage. By inducing a reversible cell cycle arrest at the G0/G1 phase through methods like serum starvation, researchers successfully suppressed post-thaw apoptosis. This intervention preserved viability, clonal growth, and immunomodulatory function at pre-freeze levels, offering a robust biochemical strategy to enhance cryopreservation outcomes [6].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for MSC Slow-Freezing Research
| Reagent / Material | Function / Explanation | Example Use |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating CPA; reduces ice crystal formation & solute damage. | Standard at 5-10% (v/v) in freezing medium. |
| Sucrose / Trehalose | Non-penetrating CPA; promotes osmotic dehydration & stabilizes membranes. | Often used at 0.1-0.3M in combination with DMSO. |
| Fetal Bovine Serum (FBS) | Provides proteins, growth factors, and lipids that membrane stability. | 10-20% (v/v) in traditional freezing media. |
| Programmable Freezer | Equipment for precise, reproducible control of cooling rate. | Essential for standardizing the -1°C/min to -3°C/min cooling rate. |
| Isopropanol Freezing Container | Passive cooling device providing an approximate -1°C/min rate. | Accessible alternative to programmable freezers. |
| Synthropic Zwitterions | Emerging CPA; inhibits ice crystallization via strong water interaction. | Used at ~10 wt% with DMSO (e.g., 15 wt%) for enhanced tissue preservation [33]. |
| Antioxidants (e.g., Catalase) | Mitigates oxidative stress from ROS generated during freeze-thaw. | Added to freezing medium to improve post-thaw function [16]. |
Workflow and Injury Mechanism Visualization
The following diagrams summarize the core experimental workflow and the physiological basis of cryoinjury during slow freezing.
Slow Freezing Workflow
Cryoinjury Mechanisms
Cryopreservation is a cornerstone of modern regenerative medicine, enabling the long-term storage of mesenchymal stem cells (MSCs) for therapeutic applications. However, conventional cryopreservation methods expose cells to various forms of cryo-injury, primarily through the formation of damaging intracellular and extracellular ice crystals [10] [34]. These crystals can mechanically disrupt cell membranes and organelles, leading to compromised viability and function post-thaw. Vitrification has emerged as a powerful technique to circumvent these challenges. It is defined as the rapid cooling of a highly concentrated cryoprotectant agent (CPA) solution,
causing it to solidify into a non-crystalline, glassy state that prevents ice formation entirely [10] [34]. While this approach effectively mitigates mechanical ice damage, it introduces other critical stresses, primarily CPA toxicity and osmotic stress, which represent significant mechanisms of cryo-injury in MSC research [10] [35]. This technical guide explores the principles, methods, and recent advancements in vitrification, framing them within the broader context of understanding and overcoming cryo-injury.
Physical Chemistry of Vitrification
The Thermodynamic Pathway to a Glassy State
The physical chemistry of vitrification is governed by the behavior of water and CPA solutions during cooling. The following diagram illustrates the critical temperature transitions and states that a solution passes through to achieve vitrification.
Critical Temperature Transitions and States in Vitrification
To achieve the glassy state depicted above, the solution must bypass the ice nucleation zones. This is accomplished through a combination of high CPA concentrations and rapid cooling rates. The CPA mixture increases the solution's viscosity dramatically, while the rapid cooling rate minimizes the time available for water molecules to organize into ice crystal nuclei [34]. The glass transition temperature (Tg) is the point at which the supercooled liquid solidifies into an amorphous glass. Above Tg, in the supercooled state, the solution remains liquid below its equilibrium freezing point but is vulnerable to both homogeneous and heterogeneous nucleation, which can lead to damaging ice crystal growth if sufficient time is provided [34]. The primary goal of vitrification protocols is to traverse this dangerous temperature zone so quickly that nucleation is effectively prevented.
Mechanisms of Cryo-injury in Vitrification
Vitrification techniques trade one form of cryo-injury (ice damage) for others:
- Cryoprotectant Toxicity (CT): The high concentrations of CPAs required for vitrification (often exceeding 6 M in total) are intrinsically toxic to cells [35]. These chemicals can disrupt cell membranes, denature proteins, and alter metabolic functions. The toxicity is exacerbated by elevated temperatures and longer exposure times, making the addition and removal of CPAs a critical and delicate phase of the protocol [10] [35].
- Osmotic Stress: During the introduction of CPAs, water rapidly exits the cell to equilibrate with the hypertonic external solution, causing cell shrinkage. Conversely, during CPA removal post-thaw, water rushes in, causing cell swelling and potential lysis. These osmotic volume changes can irreversibly damage the cell's plasma membrane and internal structures [10] [34].
Vitrification Methods and Technologies
Two primary methodological approaches exist for vitrifying biological samples, each with distinct procedural steps.
Methodological Approaches
- Equilibrium Vitrification: This method emphasizes a controlled, step-wise addition of CPA solutions, allowing the cells to gradually dehydrate and reach osmotic equilibrium with the CPA mixture before the final rapid cooling step. This careful approach minimizes osmotic shock but results in longer exposure to potentially toxic CPAs [10].
- Non-Equilibrium Vitrification: This approach prioritizes speed, using very high CPA concentrations and minimal equilibration times. The cells are rapidly exposed to the final vitrification solution and immediately plunged into liquid nitrogen. This reduces exposure time but maximizes osmotic stress and CPA toxicity, requiring extremely high cooling rates to be successful [10].
Technological Platforms for Enhanced Heat Transfer
Achieving the critical cooling rates for vitrification with lower CPA concentrations relies on technologies that maximize heat transfer.
- Convection-Based Methods: Direct immersion of samples in liquid nitrogen is common but is limited by the Leidenfrost effect, where a vapor film insulates the sample and drastically reduces the cooling rate [34].
- Conduction-Based Methods: These methods, such as the Cryotop and other solid-surface vitrification devices, press a small sample volume into a thin film or droplet onto a pre-cooled metal surface. This avoids the Leidenfrost effect and achieves very high cooling rates (>20,000°C/min) [36] [34]. Recent innovations include devices that cool from two sides, further improving heat transfer efficiency and allowing for a reduction in CPA concentration [34].
Experimental Protocols for MSC Vitrification
Standard Vitrification Protocol for 2D-Cultured MSCs
This protocol is adapted from established vitrification methods for adherent cells [10].
Materials:
- Cell Line: Human MSCs (e.g., bone marrow or umbilical cord-derived).
- Base Medium: e.g., α-MEM.
- CPA Cocktail: A typical vitrification solution may contain 5-6 M total permeating CPAs, such as 2.5-3.0 M DMSO, 2.5-3.0 M Ethylene Glycol, and 0.5-1.0 M Sucrose in base medium [10] [35].
- Device: Cryotop or similar solid-surface cooling device.
- Liquid Nitrogen.
Procedure:
- Preparation: Culture MSCs to 80-90% confluence. Prepare dilution solutions of the CPA (e.g., 10%, 20% v/v in medium) and the full-strength vitrification solution. Keep all solutions at room temperature or 4°C to reduce CPA toxicity.
- CPA Equilibration: Detach MSCs using a standard method (e.g., trypsin) and centrifuge to form a pellet.
- Resuspend the cell pellet in a low concentration CPA solution (e.g., 10%) for 3-5 minutes.
- Centrifuge and resuspend in a higher concentration (e.g., 20%) for another 3-5 minutes.
- This stepwise addition mitigates osmotic shock.
- Final Loading: Resuspend the cell pellet in the full-strength, ice-cold vitrification solution. The total exposure time in the final solution should be minimized (typically 60-90 seconds).
- Loading and Cooling: Quickly place a small volume (1-2 µL) of the cell suspension onto the Cryotop strip. Immediately plunge the device directly into liquid nitrogen. The thin film and direct contact with the cold surface achieve the required rapid cooling.
- Storage: Transfer the vitrified samples to a long-term liquid nitrogen storage tank.
- Warning: Thawing must be equally rapid to prevent ice crystallization during warming.
- Rapidly transfer the Cryotop strip into a pre-warmed (37°C) thawing solution containing 1.0 M sucrose (an osmotic buffer).
- Gently agitate for 1 minute.
- CPA Removal: To prevent osmotic shock, serially dilute the CPAs by transferring the cells through a series of solutions with decreasing sucrose concentrations (e.g., 0.5 M, 0.25 M, 0 M) every 3-5 minutes.
- Assessment: Centrifuge the cells to remove the sucrose solution, resuspend in fresh culture medium, and assess viability (e.g., via trypan blue exclusion) and functionality (e.g., differentiation potential).
Advanced Protocol: Microfluidic Vitrification of 3D-MSCs in GelMA Hydrogel
A recent innovative protocol demonstrates how physical encapsulation can improve cryosurvival and reduce CPA requirements [36]. The following workflow outlines the key steps of this advanced method.
Workflow for 3D-MSC Hydrogel Microsphere Vitrification
Materials:
- Cells: Human Umbilical Cord MSCs (hUC-MSCs).
- Hydrogel: Gelatin Methacryloyl (GelMA).
- Microfluidic Device: For generating uniform microspheres.
- CPA Solution: A reduced-concentration formulation, e.g., 15-20% lower than standard protocols [36].
Procedure:
- 3D Culture: Expand hUC-MSCs in 3D culture systems to form spheroids or clusters.
- Encapsulation: Use a microfluidic device to mix the 3D-MSC suspension with a GelMA prepolymer solution, generating uniform 3D-MSCs Hydrogel Microspheres (3D-MSCsHM). Photocrosslink the microspheres to solidify the hydrogel.
- CPA Loading: Equilibrate the 3D-MSCsHM in a reduced-CPA vitrification solution. The hydrogel matrix acts as a protective scaffold, moderating osmotic shifts.
- Vitrification and Storage: Plunge the CPA-loaded microspheres into liquid nitrogen for storage.
- Rewarming and Removal: Rapidly thaw the microspheres in a 37°C water bath and serially remove the CPAs using sucrose dilution steps.
- Functional Assessment: The study reported 96% post-thaw viability and demonstrated that the vitrified cells retained high mitochondrial integrity, metabolic function, and the capacity to promote wound healing in a mouse model, comparable to fresh 3D-MSCs [36].
Quantitative Data and Analysis
Comparative Analysis of Vitrification Solutions
The table below summarizes key performance data for different CPA formulations and techniques used in MSC vitrification.
Table 1: Performance Metrics of Vitrification Strategies for MSCs
| Vitrification Strategy | CPA Formulation | Cooling Rate | Post-Thaw Viability | Key Functional Outcome | Reference |
|---|---|---|---|---|---|
| Standard (Cryotop) | ~6-8 M total CPAs (e.g., DMSO, EG, Sugars) | >20,000 °C/min | ~70-80% | Maintains differentiation potential | [10] |
| 3D-MSCs in GelMA | Reduced CPA (∼25% less than standard) | Conduction-based | 96% | Preserved mitochondrial function & in vivo wound healing efficacy | [36] |
| M22 Solution (Research) | 9.4 M (DMSO, Formamide, EG, etc.) | Controlled slow cooling (for organs) | N/A for MSCs (Toxicity studied in ESCs) | High toxicity at 37°C; used for mutant selection | [35] |
Cryoprotectant Toxicity and Genetic Resistance
Research into the fundamental mechanisms of cryo-injury has identified specific genetic pathways linked to CPA toxicity. A forward genetic screen in mouse embryonic stem cells (ESCs) identified several mutants conferring resistance to the vitrification solution M22 [35].
Table 2: Genetic Mutants Conferring Cryoprotectant Toxicity Resistance (CTR)
| Identified Gene | Potential Pathway/Function | Observed Resistance |
|---|---|---|
| Myh9 | Non-muscle myosin, cytoskeletal organization | Resistant to M22 and Me₂SO |
| Opa1 | Mitochondrial fusion and function | Improved post-thaw survival |
| Pim1 | Serine/threonine kinase, cell survival | Resistant to M22 and Me₂SO |
| Hes1 | Notch signaling pathway | Resistant to M22 and Me₂SO |
| Ywhag | 14-3-3 protein, signal transduction | Resistant to M22 |
These mutants not only showed resistance to M22 but also to DMSO, and several demonstrated significantly improved survival after standard freezing and thawing in 10% DMSO [35]. This provides direct evidence that CT can be mitigated by specific molecular interventions, opening avenues for pharmacological blockade of CT or the engineering of more resilient MSC lines.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for MSC Vitrification Research
| Item | Function/Description | Example Application |
|---|---|---|
| Permeating CPAs (DMSO, EG) | Small molecules that cross the cell membrane, providing intracellular cryoprotection via colligative effects. | Core components of most vitrification solutions. DMSO is the most common but has known toxicity [10] [37]. |
| Non-Permeating CPAs (Sucrose, Trehalose) | Large molecules that remain outside the cell, providing extracellular protection and mitigating osmotic shock during CPA addition/removal. | Used as osmotic buffers in dilution and thawing solutions [10] [37]. |
| GelMA Hydrogel | A photocrosslinkable biomaterial that provides a protective 3D scaffold for cells during cryopreservation. | Used for encapsulating 3D-MSCs to enable vitrification with lower CPA concentrations [36]. |
| M22 Vitrification Solution | A complex, multi-component research solution for organ vitrification. Used to study fundamental mechanisms of CPA toxicity. | Tool for selecting CTR mutants in research settings [35]. |
| Controlled-Rate Freezer (CRF) | Equipment that precisely controls cooling rates, often used for protocol development and optimization before transitioning to ultra-rapid methods. | Standardizing freezing profiles for specific MSC types and container formats [38]. |
| Solid-Surface Vitrification Device | Devices like the Cryotop that enable ultra-rapid cooling via direct contact with a pre-cooled metal surface. | Standard protocol for vitrifying small cell samples like MSC clusters [10] [36]. |
Vitrification presents a powerful solution to the problem of ice-induced cryo-injury in MSC cryopreservation. The successful implementation of these techniques requires a deep understanding of the associated challenges, primarily CPA toxicity and osmotic stress. Current research is focused on developing innovative strategies to mitigate these secondary injuries, as evidenced by advanced biomaterial encapsulation techniques that allow for a significant reduction in CPA concentration while maintaining high post-thaw viability and function [36]. Furthermore, fundamental research into the genetic basis of cryoprotectant toxicity opens up new possibilities for engineering more resilient cell lines [35]. As these technologies mature, vitrification will continue to be a critical enabling technology for the clinical translation and widespread availability of MSC-based therapies.
Cryopreservation is a fundamental technique in biomedical research and clinical therapy, enabling the long-term storage of living cells, including Mesenchymal Stem Cells (MSCs). For MSCs, which are pivotal in regenerative medicine, immune therapy, and treatment of conditions like hematological diseases and COVID-19, effective cryopreservation is essential to maintain a readily available, phenotypically stable source of cells without the phenotypic drift and epigenetic alterations associated with continuous passaging [10]. The process involves cooling cells to sub-zero temperatures (typically -196°C in liquid nitrogen) to halt all metabolic activity. However, the freezing and thawing processes themselves can induce significant damage, known as cryoinjury, which compromises cell viability, function, and therapeutic potential [39] [10].
The primary mechanisms of cryoinjury in MSCs include:
- Intracellular Ice Formation (IIF): During slow cooling, water exits the cell, causing osmotic dehydration. If cooling is too rapid, water does not have time to leave the cell, leading to lethal intracellular ice crystals that disrupt organelles and the plasma membrane [39] [40].
- Solution Effects: As water freezes, solutes in the extracellular solution become concentrated, creating a hypertonic environment. This induces osmotic shock, damaging membrane lipids and proteins, and can lead to protein denaturation [39] [40].
- Oxidative Stress: The freeze-thaw cycle can generate reactive oxygen species (ROS), leading to lipid peroxidation of cell membranes, DNA damage, and activation of cell death pathways such as apoptosis [39].
- Phase Transitions: Membrane lipids can undergo damaging phase transitions during temperature changes, affecting membrane fluidity and integrity [41].
- Cell Cycle-Specific Vulnerability: Recent research has identified that MSCs in the S phase of the cell cycle are exquisitely sensitive to cryoinjury, demonstrating heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function due to double-stranded DNA breaks formed during freezing and thawing [6].
Cryoprotectant Agents (CPAs) are fundamental components added to cryopreservation media to mitigate these cryoinjury pathways. They are systematically categorized based on their ability to cross the cell membrane into Penetrating and Non-Penetrating agents, each with distinct mechanisms of action and applications [39].
Penetrating Cryoprotectants
Penetrating CPAs are low molecular weight, non-ionic molecules that readily cross the plasma membrane and enter the intracellular compartment. Their primary mechanism of action involves colligatively reducing the freezing point of water both inside and outside the cell, thereby minimizing the amount of ice formed at any given temperature. They also reduce the concentration of electrolytes in the unfrozen fraction, lessening "solution effects" injury [39] [42]. By permeating the cell, they help to stabilize intracellular proteins and prevent excessive dehydration during slow freezing [39]. However, their penetration is also the source of their primary drawback: concentration-dependent cytotoxicity.
Dimethyl Sulfoxide (DMSO)
DMSO is the most widely used penetrating CPA for many cell types, including stem cells.
- Mechanism: DMSO rapidly penetrates the cell membrane, forming hydrogen bonds with water molecules. This depresses the freezing point of water and reduces the formation of intracellular ice. It also increases the solution's viscosity, facilitating vitrification (the transition into a glassy, non-crystalline state) at high concentrations [40] [42].
- Typical Working Concentration: 5-10% (v/v) is standard for many cell types, including MSCs [10] [42].
- Cytotoxicity and Clinical Concerns: DMSO toxicity is a major concern in clinical applications. Its cytotoxicity arises from membrane effects, alterations in cell permeability, and interactions with cellular proteins and mitochondrial function [39]. Upon infusion into patients, DMSO can cause adverse reactions including nausea, vomiting, abdominal cramping, hypotension, bradycardia, and, in severe cases, cardiac arrhythmias or neurotoxicity [42]. It has also been shown to induce drastic changes in cellular processes and the epigenetic landscape in vitro [40]. Consequently, there is a strong drive to reduce or eliminate DMSO from clinical-grade cryopreservation protocols.
Glycerol
Glycerol is another common penetrating CPA, historically one of the first discovered.
- Mechanism: Similar to DMSO, glycerol lowers the freezing point of water and binds metallic ions, reducing the electrolyte concentration in the unfrozen portion. It replaces intracellular water, thereby reducing the volume of water available to form ice [43].
- Typical Working Concentration: 6-10% (v/v) is common, though optimal concentrations are cell-type specific [43].
- Cytotoxicity and Limitations: Glycerol is generally considered less toxic than DMSO but can exhibit a "contraceptive effect" in some species' spermatozoa, likely due to osmotic shock following its rapid efflux post-thaw [43]. Its larger molecular size compared to DMSO results in slower penetration across some cell membranes, which can be a limitation in certain applications [43].
Table 1: Key Characteristics of Common Penetrating CPAs
| Cryoprotectant | Molecular Weight (g/mol) | Key Mechanism of Action | Typical Working Concentration | Primary Advantages | Primary Disadvantages |
|---|---|---|---|---|---|
| DMSO | 78.1 | Depresses freezing point, increases viscosity, vitrification agent [40] [42]. | 5-10% (v/v) [10] [42] | Rapid penetration, highly effective cryoprotection [39] [42] | Significant cytotoxicity; patient side effects upon infusion [39] [42] |
| Glycerol | 92.1 | Lowers freezing point, replaces intracellular water, reduces electrolyte concentration [43]. | 6-10% (v/v) [43] | Lower general toxicity compared to DMSO [43] | Slower penetration; can cause post-thaw osmotic shock [43] |
Non-Penetrating Cryoprotectants
Non-penetrating CPAs are typically larger molecules that cannot cross the intact plasma membrane. They remain in the extracellular space and exert their protective effects through different physical mechanisms. They are generally considered less cytotoxic than penetrating CPAs and are often used in combination with them to reduce the required concentration of the toxic penetrants [44].
Sucrose
Sucrose is a disaccharide widely used as a non-penetrating supplement.
- Mechanism: Sucrose acts primarily by inducing osmotic dehydration of the cell prior to freezing. This reduces the amount of freezable intracellular water, thereby minimizing intracellular ice formation. It also increases the viscosity of the extracellular solution, supporting vitrification, and helps to mitigate osmotic shock during the thawing process when added to warming media [44].
- Typical Working Concentration: Commonly used in concentrations ranging from 0.1 M to 0.5 M in both vitrification and warming solutions [44].
Trehalose
Trehalose is a non-reducing disaccharide composed of two glucose molecules. It is a particularly interesting CPA due to its role in nature, where it protects various organisms from freezing and desiccation [41].
- Mechanism: Trehalose's action is explained by two key hypotheses:
- The Vitrification Hypothesis: Trehalose forms a high-viscosity, glassy state upon concentration (via dehydration), which prevents the molecular mobility required for ice crystal growth and recrystallization [41].
- The Water Replacement Hypothesis: Trehalose stabilizes membranes and proteins by directly hydrogen-bonding to phospholipid head groups and polar residues, effectively replacing the shell of water molecules that is lost during freezing or drying. This maintains the structural integrity of these macromolecules [41].
- Typical Working Concentration: An optimal concentration range is often observed between 100 mM to 400 mM (approximately 3.4% to 13.7% w/v). Higher concentrations can become detrimental due to excessive osmotic stress [41].
- Key Challenge and Advanced Solutions: A major limitation of trehalose is its inability to cross the cell membrane, preventing it from providing direct intracellular protection. Advanced chemical and engineering strategies are being developed to overcome this, including:
- Encapsulation: Loading trehalose into cells via endocytosis using nano- or micro-carriers.
- Cell-Penetrating Peptides (CPPs): Conjugating trehalose to peptides that facilitate its transport across the membrane.
- Engineering Membrane Permeability: Modifying the structure of trehalose or using physical methods to transiently permeabilize the membrane for trehalose loading [41].
Table 2: Key Characteristics of Common Non-Penetrating CPAs
| Cryoprotectant | Molecular Weight (g/mol) | Key Mechanism of Action | Typical Working Concentration | Primary Advantages | Primary Disadvantages |
|---|---|---|---|---|---|
| Sucrose | 342.3 | Osmotic dehydration; increases extracellular viscosity [44]. | 0.1 - 0.5 M [44] | Biocompatible, reduces osmotic shock during thawing [44] | Can cause excessive dehydration at high concentrations [44] |
| Trehalose | 342.3 | Vitrification; water replacement for membranes/proteins [41]. | 100 - 400 mM [41] | Powerful stabilizer, inspired by nature, low toxicity | Very low membrane permeability requires special delivery methods [41] |
Experimental Protocols and Data
Detailed Methodology: Synergistic CPA Combination Study
A seminal study investigating the synergism between glycerol and DMSO in buffalo spermatozoa provides a robust experimental template for CPA combination strategies [43].
Objective: To devise a cryoprotection synergism between glycerol and DMSO and to reduce the level of glycerol in the cryodiluent for buffalo spermatozoa.
Experimental Groups:
- Control: 7% glycerol.
- Group 1: 3.5% DMSO.
- Group 2: 3.5% glycerol (at 37°C) + 3.5% DMSO (at 4°C).
- Group 3: 3.5% DMSO (at 37°C) + 3.5% glycerol (at 4°C).
- Group 4 (Synergism): 1.75% glycerol + 1.75% DMSO at both 37°C and 4°C.
Processing Workflow:
- Semen Collection and Initial Evaluation: Semen was collected and evaluated for motility (≥60%) and concentration (≥500 × 10⁶/ml).
- Dilution and Cooling: Semen aliquots were diluted according to their respective group protocols, cooled from 37°C to 4°C over 2 hours, and equilibrated at 4°C for 4 hours.
- Freezing: Samples were loaded into straws and frozen in a programmable freezer using an ultra-fast freezing rate protocol.
- Storage and Thawing: Straws were stored in liquid nitrogen and later thawed in a 37°C water bath for 30 seconds.
- Post-Thaw Analysis: Sperm quality was assessed using Computer-Assisted Sperm Analysis (CASA) for motility and velocity, alongside assays for membrane integrity, mitochondrial transmembrane potential, acrosome integrity, DNA integrity, and in vivo fertility.
Key Findings: Group 4 (1.75% glycerol + 1.75% DMSO) demonstrated significantly higher post-thaw sperm quality across all measured parameters, including progressive motility, membrane integrity, mitochondrial function, and DNA integrity, compared to all other groups and the control. Crucially, the in vivo fertility rate was significantly higher for Group 4 (69.45%) compared to the control (59.81%), validating the physiological relevance of the synergistic combination [43].
Table 3: Summary of Quantitative Post-Thaw Outcomes from Synergistic CPA Study [43]
| Evaluated Parameter | Control (7% Glycerol) | Group 4 (1.75% Glycerol + 1.75% DMSO) | Statistical Significance (P-value) |
|---|---|---|---|
| Progressive Motility (%) | Lower | Higher | < 0.05 |
| Rapid Velocity (%) | Lower | Higher | < 0.05 |
| Membrane Integrity (%) | Lower | Higher | < 0.05 |
| Mitochondrial Potential (%) | Lower | Higher | < 0.05 |
| Viable Sperm with Intact Acrosome (%) | Lower | Higher | < 0.05 |
| DNA Integrity (%) | Lower | Higher | < 0.05 |
| In Vivo Fertility (%) | 59.81 | 69.45 | < 0.05 |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for CPA Research in MSC Cryopreservation
| Reagent / Material | Function / Application | Specific Example |
|---|---|---|
| Permeating CPAs (DMSO, Glycerol) | Primary intracellular cryoprotection; often used as baseline controls. | DMSO (Cell Culture Grade), Glycerol (ACS Grade) [39] [42] |
| Non-Penetrating CPAs (Sucrose, Trehalose) | Extracellular cryoprotection; used as supplements to reduce penetrating CPA concentration. | Trehalose (Pharmaceutical Grade), Sucrose (Molecular Biology Grade) [41] [44] |
| Serum / Platelet Lysate | Base component of freezing media; provides proteins and growth factors that enhance cell stability. | Fetal Bovine Serum (FBS), Human Platelet Lysate (hPL) [6] |
| Programmable Freezer | Equipment for controlled-rate slow freezing, ensuring reproducible cooling rates. | Planer Kryo 550 series or equivalent [43] |
| Liquid Nitrogen Storage System | Long-term storage of cryopreserved samples at -196°C. | Liquid nitrogen vapor-phase freezers to minimize contamination risk [10] [42] |
| Cell Viability/Cytotoxicity Assay | Quantifying post-thaw survival and CPA toxicity (e.g., apoptosis, necrosis). | Flow cytometry assays for Annexin V/PI; MTT assay for metabolic activity [6] [10] |
| Differentiation & Function Assay Kits | Confirming retention of MSC multipotency and immunomodulatory function post-thaw. | Osteogenic, adipogenic, chondrogenic differentiation kits; T-cell suppression assays [6] [10] |
Emerging Trends and Advanced Concepts
Cell Cycle Synchronization to Mitigate Cryoinjury
A groundbreaking discovery identified that MSCs in the S phase are highly vulnerable to cryoinjury, exhibiting heightened delayed apoptosis and reduced function due to DNA double-stranded breaks (DSBs) formed during freezing [6]. A mitigation strategy was developed using growth factor deprivation (serum starvation) to synchronize the cell cycle at the G0/G1 phase prior to freezing.
Experimental Workflow:
- Pre-treatment: MSCs are cultured in serum-free medium (e.g., without FBS or hPL) for 24-48 hours.
- Validation: Cell cycle analysis (e.g., via flow cytometry) confirms accumulation in G0/G1.
- Cryopreservation: Synchronized cells are cryopreserved using standard protocols (e.g., slow freezing with 10% DMSO).
- Post-Thaw Analysis: Cells are assessed for viability, clonal growth, DNA damage (γH2AX staining for DSBs), and immunomodulatory function (T-cell suppression).
Findings: This pre-treatment greatly reduced post-thaw dysfunction by preventing apoptosis linked to DNA damage in replicating cells. Viability, clonal growth, and T-cell suppression function were preserved at pre-cryopreservation levels, offering a robust, pharmacological-free strategy to enhance the recovery of therapeutic MSCs [6].
Bio-inspired and Novel CPA Strategies
Research is increasingly focused on finding biocompatible alternatives to traditional CPAs.
- Antifreeze Proteins (AFPs): Naturally occurring proteins in extremophiles that inhibit ice recrystallization through an "adsorption-inhibition" mechanism, preventing small ice crystals from growing into larger, damaging ones [39] [45].
- Natural Deep Eutectic Systems (NADES): These are supramolecular complexes of natural compounds (e.g., choline chloride and urea) that form a liquid with a depressed freezing point. They show excellent capacity for freezing point depression and ice recrystallization inhibition due to their temperature-responsive hydrogen-bonding networks [39] [45].
- Synthetic Polymers and Nanomaterials: Polymers like polyvinyl alcohol and polyampholytes act as non-penetrating CPAs by inhibiting ice recrystallization and interacting with the cell membrane. Emerging approaches also use magnetic nanoparticles for inductive heating to enable ultra-rapid and uniform warming, which is critical for avoiding devitrification (ice formation during warming) in vitrified samples [39] [40].
Visualizing Core Concepts and Workflows
Diagram: Mechanisms of Cryoinjury and CPA Protection
Diagram Title: Cryoinjury Mechanisms and CPA Protection Pathways
Diagram: Experimental Protocol for Synergistic CPA Testing
Diagram Title: Workflow for Testing Synergistic CPA Combinations
The cryopreservation of MSCs presents a complex interplay between the necessity for long-term storage and the imperative to maintain high cell viability and function post-thaw. This deep dive elucidates that the classical dichotomy between penetrating (DMSO, Glycerol) and non-penetrating (Sucrose, Trehalose) CPAs is not a matter of selecting one over the other, but rather of strategic combination. The future of CPA formulation lies in synergistic cocktails that leverage the intracellular protection of low-toxicity penetrants with the extracellular stabilization and osmotic control provided by non-penetrating agents like trehalose. Furthermore, emerging strategies such as cell cycle synchronization prior to freezing address fundamental biological vulnerabilities, moving beyond purely chemical protection. As research advances, the integration of bio-inspired molecules (AFPs, NADES), novel materials, and a deeper understanding of cell-specific stress responses will continue to refine cryopreservation protocols, ensuring that cryopreserved MSCs meet the stringent demands of clinical therapy and regenerative medicine.
Standardized Protocols and Media Formulations for Clinical-Grade MSCs
The advancement of Mesenchymal Stem/Stromal Cell (MSC)-based therapies from research to clinical application faces significant challenges, with cryopreservation representing a critical bottleneck in the cellular therapy supply chain. Effective cryopreservation ensures the stability, availability, and coordinated administration of MSC products while allowing completion of essential safety and quality control testing [46]. The mechanisms of cryo-injury during freeze-thaw cycles—particularly damage to cell membranes, the actin cytoskeleton, and labile replicating DNA—directly impact post-thaw viability, recovery, and therapeutic functionality [46] [6]. This technical guide provides a comprehensive overview of standardized protocols and media formulations for clinical-grade MSCs, framed within current understanding of cryo-injury mechanisms and their mitigation.
Standardization following Good Manufacturing Practices (GMP) is essential for clinical translation. Protocols must ensure batch-to-batch consistency, product safety, and maintain critical quality attributes including viability, identity, potency, and sterility [47]. The International Society for Cellular Therapy (ISCT) establishes minimal criteria for defining MSCs: plastic-adherence, specific surface marker expression (CD73, CD90, CD105 ≥95%; CD45, CD34, CD14/CD11b, CD79α/CD19, HLA-DR ≤2%), and tri-lineage differentiation potential [10]. This guide details the current landscape of cryopreservation methodologies, media formulations, and mechanistic insights into cryo-injury, providing researchers and drug development professionals with actionable frameworks for clinical-grade MSC production.
Understanding Cryo-Injury: Molecular Mechanisms and Implications
Fundamental Cryo-Injury Pathways in MSCs
Cryo-injury occurs through multiple interconnected mechanisms during freezing and thawing. Intracellular ice formation physically damages membranes and organelles, while osmotic stress from solute concentration changes causes deleterious cell volume fluctuations [10]. Recent research has identified a previously underappreciated mechanism: DNA double-stranded breaks (DSBs) in replicating cells.
A 2023 study revealed that S-phase MSCs are exquisitely sensitive to cryopreservation-induced damage. The cryopreservation process induces DSBs in labile replicating DNA, triggering delayed apoptosis post-thaw and significantly reducing immunomodulatory function [6]. This cell cycle-dependent vulnerability explains the variable post-thaw recovery observed in heterogeneous MSC populations and presents a novel target for intervention.
Consequences of Cryo-Injury on Therapeutic Potential
The functional consequences of cryo-injury extend beyond immediate cell death. Even in surviving cells, damage can manifest as:
- Reduced clonal growth and proliferative capacity [6]
- Impaired immunomodulatory function, particularly T-cell suppression capability [6]
- Altered secretory profiles affecting paracrine-mediated therapeutic effects [1]
- Changes in transcriptional and gene expression profiles that may impact in vivo performance [46]
Understanding these mechanisms enables the development of targeted strategies to mitigate cryo-injury, rather than relying solely on empirical optimization of cryopreservation formulas.
Cryopreservation Media Formulations: Composition and Performance
Cryoprotective Agents (CPAs) are essential components of cryopreservation media, categorized as penetrating (e.g., DMSO, glycerol) or non-penetrating (e.g., sucrose, trehalose). They function collectively to minimize ice crystal formation, stabilize cell membranes, and manage osmotic stress during phase transitions [46] [10].
Table 1: Comparative Analysis of Clinical-Grade Cryopreservation Media Formulations
| Formulation Type | Key Components | Reported Post-Thaw Viability | Recovery of Viable MSCs | Advantages | Limitations |
|---|---|---|---|---|---|
| Traditional DMSO-based | 5-10% DMSO in plasma-lyte or saline [46] | 89.8% (95% CI: 84.9-94.7%) [46] | 87.3% (95% CI: 82.1-92.5%) [46] | Established protocol, high efficacy | DMSO-related toxicity concerns [4] |
| Novel DMSO-free (SGI) | Sucrose, glycerol, isoleucine in Plasmalyte A [46] | 82.9% (95% CI: 76.0-89.8%) [46] | 92.9% (95% CI: 85.7-100.0%) [46] | Eliminates DMSO toxicity, better recovery | Slightly reduced viability |
| Low-DMSO with rHSA | 1-5% DMSO + recombinant Human Serum Albumin [48] | Maintained at 1-2.5% DMSO [48] | Improved vs. no-albumin controls [48] | Reduced DMSO exposure, enhanced recovery | Requires optimization for different cell types |
| Commercial DMSO-free (PRIME-XV) | Proprietary non-penetrating CPAs [49] | Validated for multiple cell types [49] | Maintains phenotype and function [49] | Regulatory-friendly, eliminates wash step | Limited public composition data |
| Clinical-Grade (CS-SC-D1) | GMP-manufactured, NMPA-approved [50] | >90% [50] | 15% improvement in cell yields [50] | Regulatory approved, source flexible | Newer product with less extensive validation |
DMSO-Containing Formulations
DMSO (Dimethyl Sulfoxide) remains the most widely used penetrating CPA in clinical cryopreservation due to its proven efficacy. However, concerns regarding potential patient toxicity—including allergic reactions, hemodynamic instability, and neurological effects—have driven efforts to reduce or eliminate DMSO from clinical products [4]. When used, concentrations of 5-10% are typical, with recent approaches successfully reducing levels to 1-2.5% when supplemented with recombinant Human Serum Albumin (rHSA) [48].
DMSO-Free Alternatives
DMSO-free formulations represent an active area of innovation, combining non-penetrating CPAs like sucrose (stabilizes membranes) and trehalose (forms glassy state during freezing) with less toxic penetrating agents like glycerol and amino acids such as isoleucine [46]. International multicenter studies demonstrate that while DMSO-free solutions may yield slightly lower viability (82.9% vs. 89.8%), they provide superior recovery of viable MSCs (92.9% vs. 87.3%) and comparable immunophenotype and global gene expression profiles [46].
Standardized Cryopreservation Methodologies
Slow Freezing: The Current Clinical Standard
Slow freezing represents the predominant method for clinical MSC cryopreservation, with controlled cooling rates allowing sufficient cellular dehydration and minimizing intracellular ice formation [10].
Diagram 1: Standard Slow Freezing Workflow (6 steps)
Critical Protocol Parameters:
- Cooling rate: -1°C/minute to -3°C/minute [10]
- CPA addition: Stepwise at 4°C to minimize osmotic shock
- Final storage: Liquid nitrogen vapor phase (-135°C to -196°C)
- Thawing: Rapid in 37°C water bath (>100°C/minute) [10]
Vitrification as an Emerging Alternative
Vitrification uses high CPA concentrations and ultra-rapid cooling to achieve a glassy state without ice crystal formation. While promising, it faces technical challenges for larger volume MSC samples, including CPA toxicity and devitrification risks during warming [10].
Mitigating Cryo-Injury Through Biological Interventions
Cell Cycle Synchronization
Addressing the mechanism of DNA damage in replicating cells, researchers have developed a pre-freezing serum starvation protocol that synchronizes MSCs in G0/G1 phase, where they are less vulnerable to cryo-injury. This intervention significantly reduces post-thaw apoptosis and preserves immunomodulatory function without requiring genetic modification or cytokine priming [6].
Diagram 2: Cryo-Injury Mechanism & Intervention (4 steps each)
CPA Removal and Post-Thaw Processing
Effective CPA removal, particularly for DMSO-containing formulations, is essential to minimize patient exposure. However, post-thaw washing introduces additional manipulation that can reduce cell yields by 5-25% through mechanical stress and centrifugation-induced apoptosis [10]. Closed-system automated washers and reduced-volume infusions represent evolving approaches to balance DMSO exposure with cell loss.
Quality Assessment and Release Criteria
Comprehensive post-thaw assessment is essential for clinical batch release. Standardized evaluation should include:
Table 2: Essential Post-Thaw Quality Control Metrics for Clinical-Grade MSCs
| Quality Attribute | Assessment Method | Acceptance Criteria | Clinical Significance |
|---|---|---|---|
| Viability | Flow cytometry with viability dyes (e.g., 7-AAD, PI) | ≥70-80% (typically >90% achieved) [46] [47] | Ensures sufficient living cells for therapeutic effect |
| Recovery | Viable cell count pre-/post-cryopreservation | ≥85% (protocol-dependent) [46] | Indicates process efficiency and scalability |
| Identity/Purity | Flow cytometry for CD73, CD90, CD105 (≥95%); CD45, CD34, HLA-DR (≤2%) [10] | ISCT criteria [10] | Confirms MSC identity and absence of contaminants |
| Potency | T-cell suppression assay; differentiation potential | Donor-dependent baseline [6] | Demonstrates functional therapeutic capacity |
| Sterility | BacT/Alert, mycoplasma testing, endotoxin | No growth; endotoxin <5 EU/kg/hr [47] | Ensures patient safety |
| Genetic Stability | Karyotyping or transcriptomic analysis | Normal profile; comparable to pre-freeze [46] | Confirms absence of cryopreservation-induced changes |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Clinical-Grade MSC Cryopreservation Research
| Reagent Category | Specific Examples | Function | Considerations for Clinical Translation |
|---|---|---|---|
| Basal Solutions | Plasmalyte A [46] | Electrolyte-balanced cryopreservation base | Prevents osmotic shock; clinically compatible |
| Penetrating CPAs | DMSO (USP grade) [46] | Prevents intracellular ice formation | Toxicity concerns driving reduction/elimination [4] |
| Non-Penetrating CPAs | Sucrose, trehalose [46] [10] | Osmotic stabilizers, membrane protectants | DMSO-free formulation components |
| Protein Stabilizers | Recombinant Human Serum Albumin (Optibumin) [48] | Membrane stabilization, reduces apoptosis | Animal-origin-free eliminates adventitious agent risk |
| Commercial Media | PRIME-XV FreezIS DMSO-Free [49] | Optimized, ready-to-use formulations | Regulatory support documentation available |
| Cell Culture Media | MSC-Brew GMP Medium [47] | Pre-freeze expansion maintaining potency | Enhanced proliferation while retaining stemness |
| Viability Assays | Flow cytometry with 7-AAD/Annexin V [6] | Apoptosis and necrosis quantification | Distinguishes immediate vs. delayed-onset cell death |
The standardization of cryopreservation protocols for clinical-grade MSCs requires careful balancing of traditional approaches with emerging innovations. While DMSO-containing formulations currently offer the most established track record, safety concerns continue to drive adoption of DMSO-free alternatives that demonstrate comparable performance in multicenter studies [46]. The integration of biological interventions like cell cycle synchronization addresses fundamental cryo-injury mechanisms, particularly DNA damage in replicating cells [6].
Future directions will likely focus on personalized cryopreservation protocols tailored to specific MSC tissue sources and clinical applications, closed-system automated processing to enhance reproducibility and minimize contamination risk, and potency-based quality metrics that better predict in vivo therapeutic efficacy. As the field advances toward increasingly standardized, GMP-compliant workflows, comprehensive understanding of both the practical protocols and underlying cryo-injury mechanisms will ensure the consistent production of high-quality MSC products capable of delivering on their therapeutic promise.
The thawing process is a critical determinant of post-preservation cell survival and functionality in mesenchymal stem cell (MSC)-based therapies. This technical guide examines the mechanisms of cryo-injury pertinent to thawing and provides evidence-based protocols for rapid warming and cryoprotectant agent (CPA) removal. When properly executed, these processes maintain MSC viability, potency, and functionality at levels comparable to fresh cells, enabling true "off-the-shelf" therapeutic applications. Conversely, suboptimal thawing can induce severe osmotic stress, membrane damage, and cytoskeletal disruption, fundamentally compromising MSC therapeutic efficacy.
Cryopreserved MSCs represent a cornerstone of regenerative medicine, offering the potential for immediate treatment of acute conditions including stroke, myocardial infarction, and retinal ischemia [51]. The freezing process inevitably introduces cellular stresses; however, the thawing process presents equally critical challenges where improper execution can negate previous preservation successes.
The fundamental mechanisms of cryo-injury during thawing include:
- Intracellular Ice Crystal Recrystallization: During slow warming, microscopic ice crystals formed during freezing can grow and merge, causing severe mechanical damage to intracellular organelles and membranes [10].
- Osmotic Shock During CPA Removal: Rapid dilution of permeating CPAs like DMSO creates steep osmotic gradients, causing excessive water influx, cell swelling, and potential membrane rupture [10] [52].
- Toxic CPA Exposure: Extended exposure to DMSO at physiological temperatures initiates dose- and time-dependent cytotoxicity, compromising membrane integrity and cellular functions [53].
Optimal thawing protocols must therefore achieve two conflicting objectives simultaneously: sufficiently rapid warming to prevent ice recrystallization while ensuring controlled CPA removal to prevent osmotic damage.
Fundamental Principles of Thawing
The Imperative of Rapid Warming
The consensus across cryopreservation literature mandates rapid warming until complete ice dissolution. The standard methodology involves transferring vials directly from liquid nitrogen storage to a 37°C water bath with gentle agitation until no ice crystals remain [10]. This rapid thermal transition (typically exceeding 100°C/min) bypasses the dangerous temperature zone (-50°C to -20°C) where ice recrystallization occurs most readily.
Water baths currently represent the gold standard for heat transfer efficiency; however, contamination risk from waterborne microorganisms presents significant concerns for clinical applications. Aseptic alternatives including dry heating devices and automated thawing systems are emerging to maintain warming rates while eliminating contamination risks [10].
CPA Removal: Balancing Toxicity and Osmotic Stress
Following rapid warming, immediate CPA removal is essential to mitigate DMSO toxicity. However, the removal process itself introduces osmotic stress as extracellular CPA concentration decreases rapidly, creating osmotic imbalance that drives water into cells. The resulting swelling can exceed critical volume limits, causing membrane rupture if not properly managed [10] [52].
Table 1: Comparative Analysis of Thawing and Reconstitution Solutions for MSCs
| Solution Composition | Post-Thaw Viability | Cell Loss After 1h | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Saline + 2% HSA | >90% | <10% | Prevents dilution-induced cell loss; clinical compatibility | Requires pharmaceutical-grade HSA |
| Protein-Free Saline | <80% | ~40-50% | Simple formulation | High instantaneous cell loss |
| Phosphate-Buffered Saline (PBS) | <80% | >40% | Standard buffer composition | Poor MSC stability |
| Culture Medium | Variable | >40% | Provides nutrients | Poor MSC stability post-thaw |
Critical Parameters in Thawing Protocol Optimization
Solution Composition and Protein Requirement
Reconstitution solution composition profoundly impacts MSC recovery. Research demonstrates that protein-free thawing solutions incur up to 50% immediate cell loss [52]. The addition of 2% human serum albumin (HSA) to isotonic saline virtually eliminates this loss, maintaining >90% viability with no significant cell loss for at least four hours post-thaw. Simple isotonic saline alone provides superior stability compared to phosphate-buffered saline (PBS) or culture medium, which both demonstrate >40% cell loss within one hour [52].
Cell Concentration Effects
Post-thaw cell concentration critically influences survival. Diluting MSCs below 1×10⁵ cells/mL in protein-free vehicles triggers instant cell loss exceeding 40% with viability dropping below 80% [52]. Optimal reconstitution maintains concentrations at approximately 5×10⁶ cells/mL to minimize adhesion-related losses and preserve membrane integrity during the critical recovery phase.
Temporal Considerations
The post-thaw recovery timeline presents competing priorities: rapid CPA removal versus functional recovery. While CPA removal should begin immediately after thawing, evidence suggests that some functional attributes require extended recovery periods:
- Viability and Metabolic Activity: Significantly reduced at 24 hours post-thaw but recover to near-baseline levels by 72 hours [51].
- Immunomodulatory Function: Preserved immediately post-thaw when viability is maintained [51].
- Cytoskeletal Organization and Engraftment: Disrupted immediately after thawing but largely restored after 48 hours of culture [51].
These temporal recovery patterns inform application-specific protocols: MSCs retain immunomodulatory potency immediately post-thaw for secretory applications, while engraftment-dependent therapies may benefit from brief recovery periods.
Experimental Protocols for Thawing Methodology
Standardized Thawing and Reconstitution Protocol
The following optimized protocol maximizes MSC recovery and functionality [52]:
- Preparation: Pre-warm water bath to 37°C. Prepare reconstitution solution (0.9% saline with 2% clinical-grade HSA).
- Rapid Warming: Transfer cryovial from liquid nitrogen to 37°C water bath with gentle agitation until no ice visible (typically 2-3 minutes).
- Initial Dilution: Transfer vial contents to 15mL conical tube. Gradually add 10mL pre-warmed reconstitution solution dropwise over 1-2 minutes with gentle agitation.
- Centrifugation: Centrifuge at 300-400×g for 5 minutes.
- Resuspension: Discard supernatant and resuspend cell pellet in appropriate administration solution at minimum 5×10⁶ cells/mL.
- Storage: Processed cells maintain viability >90% for up to 4 hours at room temperature in optimized solution.
Assessment Methodology for Post-Thaw MSC Quality
- Viability Assessment:
- 7-AAD/Annexin V Staining: Standard flow cytometry apoptosis/necrosis detection.
- TUNEL Assay: Detects DNA fragmentation; reveals minimal cell death when properly executed [51].
- Functionality Assessment:
- Immunomodulatory Potency: Co-culture with CD3/CD28-stimulated PBMCs; suppression of proliferation indicates maintained function [51].
- IDO Activity: Kynurenine production measurement after IFN-γ stimulation [51] [54].
- In Vivo Efficacy: Retinal ischemia/reperfusion model demonstrates cryopreserved MSC efficacy equivalent to fresh cells [51].
Diagram 1: MSC Thawing Process Workflow and Critical Control Points. Optimal pathway (yellow/green) contrasts with suboptimal outcomes (red) resulting from procedural deviations.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Essential Research Reagents for MSC Thawing Protocols
| Reagent/Material | Function/Purpose | Specifications/Alternatives |
|---|---|---|
| Human Serum Albumin (HSA) | Prevents cell loss during thawing and dilution; stabilizes membranes | Clinical-grade, 2% in isotonic saline |
| DMSO Cryoprotectant | Permeating CPA requiring removal post-thaw | Typically 10% concentration; cytotoxic at room temperature |
| Isotonic Saline (0.9%) | Base solution for reconstitution | Physiological osmolarity; superior to PBS for post-thaw stability |
| Water Bath | Provides rapid, uniform heating | 37°C calibrated; contamination risk necessitates sealed vial containers |
| Clinical-Grade Culture Medium | Post-thaw recovery medium when immediate use isn't required | Contains serum or platelet lysate for cell adhesion and spreading |
| Interferon-γ (IFN-γ) | Priming agent to enhance immunomodulatory potency | Pre-treatment pre-cryopreservation improves post-thaw function [54] |
The thawing process represents a critical juncture in MSC cryopreservation where controlled rapid warming and strategic CPA removal determine therapeutic cell quality. Optimal protocols employing protein-containing isotonic solutions and maintaining appropriate cell concentrations preserve viability and functionality. When properly executed, cryopreserved MSCs maintain immunomodulatory potency, secretory function, and in vivo efficacy comparable to fresh cells, enabling true "off-the-shelf" availability for regenerative applications. Standardization of these protocols across laboratories and clinical trials will enhance reproducibility and accelerate the translational progress of MSC-based therapies.
Minimizing Cryodamage: Advanced Strategies for Protocol Optimization and Problem-Solving
The clinical development of mesenchymal stromal cell (MSC)-based therapies faces a significant logistical challenge: the need for effective cryopreservation that maintains post-thaw cell viability, potency, and function. Cryopreservation enables the generation of "off-the-shelf" cellular therapies that can be distributed and stored for immediate use in treating acute conditions. However, conventional cryopreservation methods using dimethyl sulfoxide (DMSO) as a cryoprotective agent (CPA) result in substantial loss of post-thaw cell viability and function, creating a major bottleneck in the regenerative medicine pipeline [55]. The "cold truth" is that cryopreservation introduces significant variability into therapeutic product development, implying substantial financial losses and potential safety concerns [55].
Understanding cryoinjury mechanisms is fundamental to developing effective mitigation strategies. Recent research has identified that S phase MSCs demonstrate exquisite sensitivity to cryoinjury, exhibiting heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function [6]. The fundamental injury mechanism involves double-stranded breaks in labile replicating DNA that form during the cryopreservation and thawing processes [6]. Beyond genetic damage, cryopreservation reduces cell viability, increases apoptosis, impairs metabolic activity, and adversely affects adhesion potential—attributes critical for MSC therapeutic efficacy [55]. These effects persist for at least 24 hours post-thaw, suggesting that common recovery periods may be insufficient for complete functional restoration [55].
Fundamental Mechanisms of Cryoinjury in MSCs
Cell Cycle-Dependent Vulnerability
Recent investigations have revealed that cryoinjury does not affect all MSCs uniformly. Cells in the S phase of the cell cycle are particularly vulnerable to cryopreservation-induced damage. During cryopreservation and thawing, these replicating cells suffer DNA double-stranded breaks (DSBs) in labile replicating DNA, leading to delayed apoptosis and significantly reduced post-thaw function [6]. This cell cycle-dependent vulnerability represents a fundamental cryoinjury mechanism that had previously been overlooked in cryopreservation protocol development.
The heightened sensitivity of S phase cells has profound implications for MSC therapy potency. When therapeutic doses contain a substantial proportion of these vulnerable cells, the overall efficacy of the treatment may be compromised even if initial viability appears acceptable. This mechanism explains why traditional approaches focusing solely on membrane integrity and immediate viability assessment have failed to ensure consistent post-thaw therapeutic potency.
Temporal Progression of Post-Thaw Dysfunction
The damage incurred during cryopreservation manifests progressively in the post-thaw period, creating a complex recovery challenge:
- Immediately post-thaw (0-4 hours): Cells exhibit reduced viability, increased apoptosis, impaired metabolic activity, and diminished adhesion potential [55]. Membrane pores formed during DMSO exposure can lead to inaccurate viability assessments with traditional markers [51].
- 24 hours post-thaw: Viability typically recovers and apoptosis levels drop, but metabolic activity and adhesion potential remain significantly lower than fresh cells [55].
- Beyond 24 hours: Effects become more variable between cell lines, with some maintaining proliferation capacity while others show reduced clonal growth and differentiation potential [55].
This temporal progression underscores the importance of long-term assessment of MSC attributes beyond the standard 24-hour recovery period, particularly for critical quality attributes that may not manifest immediately.
Strategic Approaches to DMSO Reduction and Replacement
CPA Mixture Optimization for Toxicity Reduction
The systematic investigation of CPA mixtures has revealed promising strategies for reducing overall toxicity while maintaining cryoprotective efficacy. High-throughput screening of 22 individual CPAs and numerous binary mixtures at concentrations up to 12 mol/kg has demonstrated that toxicity neutralization is achievable through careful formulation [56].
Table 1: Effective CPA Combinations Demonstrating Toxicity Reduction
| CPA Combination | Total Concentration | Viability Outcome | Temperature |
|---|---|---|---|
| Formamide + Glycerol | 12 mol/kg | 97% viability (vs. 20% with formamide alone) | 4°C |
| Various binary mixtures | 6 mol/kg | 12 combinations with significantly reduced toxicity | 4°C |
| Various binary mixtures | 12 mol/kg | 8 combinations with significantly reduced toxicity | 4°C |
Notably, at 4°C—a temperature commonly used for CPA equilibration in tissue and organ cryopreservation—CPA toxicity is significantly reduced compared to room temperature [56]. This temperature effect provides an additional parameter for optimization in CPA formulation development. Particularly promising is the observation that certain combinations, especially those involving formamide, acetamide, dimethyl sulfoxide, and glycerol, can neutralize the toxicity of individual components [56].
Emerging Alternative Cryoprotective Agents
Deep Eutectic Solvents (DES)
Deep eutectic solvents represent a novel class of cryoprotective agents with tunable composition, low toxicity, and favorable biocompatibility. These systems are formed by combining a hydrogen-bond donor and acceptor to yield a eutectic mixture with a depressed melting point [57]. Their extensive hydrogen-bond networks confer strong solvency for biomolecules and contribute to membrane and protein stabilization—properties essential for effective cryopreservation.
In platelet cryopreservation, the addition of 10% choline chloride-glycerol DES to DMSO-free protocols demonstrated excellent post-thaw recovery (>85%) and maintained functional integrity comparable to control conditions [57]. This suggests that DES formulations may offer a viable path toward reducing or eliminating DMSO from cryopreservation protocols while maintaining post-thaw cell quality.
Controlled Rate Freezing with Minimal CPAs
An emerging approach eliminates DMSO entirely through optimized physical parameters rather than chemical protection. The combination of controlled rate freezing (CRF) with minimal cryoprotectants demonstrates that satisfactory post-thaw recovery is achievable with substantially reduced CPA exposure [57]. For platelets, CRF with only NaCl maintained post-thaw recovery of 86.9±0.1% without DMSO [57], challenging the convention that high CPA concentrations are necessary for effective cryopreservation.
Novel Mitigation Strategies Beyond CPA Formulation
Cell Cycle Synchronization
A paradigm-shifting approach to mitigating cryoinjury involves preparing cells for cryopreservation through cell cycle synchronization. By blocking cell cycle progression at G0/G1 through growth factor deprivation (serum starvation), researchers successfully prevented apoptosis induced by double-stranded breaks in labile replicating DNA [6]. This intervention:
- Greatly reduced post-thaw dysfunction of MSC
- Preserved viability, clonal growth, and T cell suppression function at pre-cryopreservation levels
- Achieved results comparable to cells prior to freezing or frozen after priming with IFNγ
This strategy represents a fundamental shift from focusing exclusively on the cryopreservation medium to preconditioning the cells themselves, addressing the root cause of S phase-associated vulnerability.
Immunomodulatory Pre-licensing
Another innovative approach involves "pre-licensing" MSCs with immunomodulatory agents prior to cryopreservation. Priming cells with potent cytokines such as interferon gamma (IFNγ) before freezing has been shown to enhance post-thaw function in some contexts [6] [7]. However, this strategy requires careful optimization, as one study demonstrated that IFNγ pre-licensed cryopreserved MSCs unexpectedly lost effectiveness in an in vivo retinal ischemia/reperfusion model, potentially due to increased MHC expression accelerating immune detection and clearance [7].
The variability in outcomes with pre-licensing strategies highlights the importance of disease-specific validation, as the appropriateness of specific preconditioning strategies cannot be determined outside the context of a particular pathology [7].
Diagram 1: Integrated workflow combining multiple cryopreservation optimization strategies to mitigate CPA toxicity and enhance post-thaw MSC function.
Quantitative Assessment of Post-Thaw MSC Function
Comprehensive Functional Metrics
Evaluating the success of cryopreservation strategies requires moving beyond simple viability measures to include comprehensive functional assessment. Multiple studies have demonstrated that viability alone is an insufficient metric for judging therapeutic potential [55] [51]. A robust assessment should include:
- Viability and apoptosis levels at multiple time points (0, 2, 4, 24 hours post-thaw)
- Metabolic activity and adhesion potential
- Immunomodulatory potency (IDO expression, kynurenine production, PBMC suppression)
- Proliferation rate and clonal growth capacity
- Differentiation potential (adiopgenic and osteogenic capacity)
- In vivo therapeutic efficacy in disease-relevant models
Table 2: Temporal Progression of Post-Thaw MSC Recovery Based on Quantitative Studies
| Time Post-Thaw | Viability & Apoptosis | Metabolic Activity | Adhesion Potential | Immunomodulatory Function |
|---|---|---|---|---|
| 0-4 hours | Reduced viability, increased apoptosis | Impaired | Impaired | Variable (donor-dependent) |
| 24 hours | Recovered viability, reduced apoptosis | Remains impaired | Remains impaired | May require 48h for full recovery |
| Beyond 24 hours | Normalized | Gradually recovers | Gradually recovers | Context-dependent efficacy |
The data clearly demonstrate that fresh and cryopreserved MSCs are biologically different, and these differences introduce variability that must be accounted for in therapeutic product development [55]. The recovery trajectory is donor-dependent and influenced by specific cryopreservation conditions, emphasizing the need for individualized optimization.
Standardized Experimental Protocols
To ensure reproducible assessment of cryopreservation outcomes, the following standardized protocols are recommended based on cited studies:
Cell Cycle Synchronization Protocol:
- Culture MSCs to 70-80% confluence
- Replace complete medium with serum-free medium for 48-72 hours
- Verify G0/G1 arrest via flow cytometry before cryopreservation
- Cryopreserve using standard protocols with optimized CPA mixtures [6]
CPA Toxicity Screening Protocol:
- Prepare CPA solutions at target concentrations (e.g., 6-12 mol/kg)
- Expose cells to CPAs for standardized duration at both room temperature and 4°C
- Assess viability using multiple methods (PI exclusion, TUNEL, metabolic assays)
- Evaluate functional retention post-thaw [56]
Comprehensive Post-Thaw Assessment Protocol:
- Thaw cells rapidly at 40°C for 1 minute
- Dilute DMSO immediately in warm complete medium
- Assess viability at 0, 2, 4, and 24 hours post-thaw
- Evaluate metabolic activity (XTT assay) at 24, 48, and 72 hours
- Test immunomodulatory function (IDO expression, PBMC suppression) at 48-72 hours
- Assess long-term functionality (proliferation, differentiation) beyond 72 hours [55] [51]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CPA Toxicity Mitigation Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Traditional CPAs | DMSO, Glycerol, Formamide, Acetamide | Baseline cryoprotection; component of toxicity-reduction mixtures |
| Emerging CPAs | Choline chloride-glycerol DES, Proline-glycerol DES | DMSO alternatives with potential reduced toxicity and enhanced biocompatibility |
| Cell Cycle Modulators | Serum-free media, Growth factor deprivation agents | Induce G0/G1 arrest to reduce S-phase vulnerability |
| Immunomodulatory Primers | Interferon-gamma (IFNγ) | Enhance immunomodulatory function pre-cryopreservation |
| Viability Assessment Tools | PI, Annexin V, TUNEL assay, XTT metabolic assay | Comprehensive viability and function assessment |
| Functional Assay Reagents | IDO ELISA kits, Kynurenine detection, PBMC suppression assay components | Evaluate immunomodulatory potency retention |
The mitigation of CPA toxicity in MSC cryopreservation requires a multifaceted approach that addresses both the chemical composition of cryoprotectant solutions and the biological state of the cells themselves. The emerging paradigm recognizes that effective cryopreservation extends beyond cell survival to encompass functional retention and therapeutic potency.
The most promising strategies include:
- Cell cycle synchronization to minimize S-phase vulnerability
- Optimized CPA mixtures that exploit toxicity neutralization effects
- Novel cryoprotectants like deep eutectic solvents with improved biocompatibility
- Physical optimization through controlled rate freezing with minimal CPAs
Future research should focus on validating these approaches in disease-specific contexts, as the appropriateness of any cryopreservation strategy is ultimately determined by its ability to maintain therapeutic efficacy for specific clinical applications [7]. The development of standardized, functionally-validated cryopreservation protocols will be essential for realizing the full potential of "off-the-shelf" MSC therapies across diverse medical conditions.
The clinical development of cellular therapies based on Mesenchymal Stem/Stromal Cells (MSCs) has been hindered by ineffective cryopreservation methods that result in substantial loss of post-thaw cell viability and function [6]. Cryopreservation represents a critical enabling technology for the MSC field, allowing for long-term storage of these living cellular resources until their therapeutic application in conditions ranging from graft-versus-host disease to orthopedic injuries and coronary artery disease [10] [58]. The fundamental challenge lies in navigating the physical phase change of water from liquid to solid while minimizing the damage that ice formation can inflict on delicate cellular structures.
The pursuit of optimized cooling protocols is not merely an academic exercise—it addresses a pressing clinical need. Post-thaw dysfunction in MSCs can manifest as reduced viability, impaired immunomodulatory function, and limited engraftment potential, ultimately compromising therapeutic efficacy [6]. Different MSC sources, including bone marrow (BM-MSCs), adipose tissue (AD-MSCs), and umbilical cord (UC-MSCs), present unique cryopreservation challenges due to variations in their size, membrane permeability, and physiological characteristics [1]. This technical guide examines the core mechanisms of cryogenic injury in MSCs and provides evidence-based strategies for optimizing cooling rates and ice nucleation control, with the goal of maximizing post-thaw recovery and functionality across diverse MSC sources.
Fundamental Mechanisms of Cryo-Injury in MSCs
Understanding the specific cellular injuries that occur during freezing is prerequisite to developing optimized protocols. Cryo-injury in MSCs manifests through several distinct but interconnected mechanisms.
Physical and Biochemical Injury Pathways
The formation of intracellular ice crystals represents one of the most devastating events during cryopreservation, directly causing mechanical damage to membranes and organelles [10]. During slow cooling, water exits cells to freeze externally, leading to excessive cell shrinkage and solution effects—harmful increases in solute concentration that denature proteins and disrupt lipid membranes [10] [59]. The process of removing cryoprotective agents (CPAs) post-thaw can also induce osmotic stress, leading to damaging cell volume fluctuations that may culminate in cell lysis [10].
Recent research has identified a previously overlooked mechanism of cryo-injury specific to replicating cells. MSC populations naturally contain cells at different cell cycle stages, and those in S phase demonstrate exquisite sensitivity to cryo-injury [6]. These cells show heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function, attributed to double-stranded breaks in labile replicating DNA that form during the cryopreservation and thawing processes [6].
Table 1: Fundamental Mechanisms of Cryo-Injury in MSCs
| Injury Mechanism | Underlying Cause | Primary Consequence |
|---|---|---|
| Intracellular Ice Formation | Inadequate dehydration before ice nucleation | Mechanical damage to membranes and organelles |
| Solution Effects | Concentration of solutes in unfrozen fraction | Protein denaturation, membrane disruption |
| Osmotic Stress | Improper CPA addition/removal | Cell shrinkage/swelling, membrane damage |
| CPA Toxicity | Chemical effects of cryoprotectants | Metabolic disruption, compromised function |
| DNA Damage | Cryopreservation of S-phase cells | Delayed apoptosis, reduced immunomodulation |
Visualizing Cryo-Injury Pathways in MSCs
The following diagram illustrates the interconnected pathways of cryo-injury in MSCs and potential intervention strategies to mitigate damage.
The optimal cooling rate for MSCs represents a balance between the competing risks of intracellular ice formation (too rapid cooling) and excessive dehydration/solute damage (too slow cooling). This balance varies significantly between MSC sources due to intrinsic biological differences.
Source-Specific Cooling Parameters
Bone marrow-derived MSCs (BM-MSCs) typically respond well to standard slow freezing protocols with cooling rates of -1°C/min, achieving approximately 70-80% viability post-thaw [10]. These established protocols make BM-MSCs relatively straightforward to cryopreserve, though optimized approaches can further enhance recovery.
Adipose tissue-derived MSCs (AD-MSCs) demonstrate similar tolerance to standard slow freezing rates but may benefit from slightly modified protocols. Their larger lipid content influences membrane fluidity and may necessitate attention to CPA permeation kinetics during protocol development.
Umbilical cord-derived MSCs (UC-MSCs) present distinct challenges due to their generally smaller size and different membrane characteristics. Some evidence suggests they may tolerate moderately faster cooling rates than BM-MSCs, though the standard -1°C/min remains widely effective [1].
Table 2: Experimentally Determined Optimal Cooling Parameters for Different MSC Sources
| MSC Source | Optimal Cooling Rate | Plunge Temperature | Post-Thaw Viability | Key Considerations |
|---|---|---|---|---|
| Bone Marrow (BM-MSC) | -1°C/min | -40°C to -50°C | 70-80% [10] | Well-established protocol |
| Adipose Tissue (AD-MSC) | -1°C/min to -2°C/min | -40°C to -50°C | 70-85% | Lipid content affects CPA permeation |
| Umbilical Cord (UC-MSC) | -1°C/min to -3°C/min | -40°C to -50°C | 75-85% | Smaller cell size may permit faster cooling |
Advanced Protocol: Interrupted Cooling Strategies
Interrupted cooling protocols, where the cooling process is temporarily halted within specific sub-zero temperature ranges, represent an advanced strategy to improve viability [60]. These methods allow researchers to study cell response during freeze/thaw processes and optimize critical factors affecting post-thaw function.
Two-Step Rapid Cooling Protocol:
- Cool samples rapidly (e.g., -10°C to -20°C/min) to an intermediate temperature between -20°C and -40°C
- Hold at this temperature for 5-15 minutes to allow for partial dehydration
- Plunge into liquid nitrogen for final cooling to -196°C
Graded Freezing Approach:
- Initiate cooling at -1°C/min to -40°C
- Transfer to a -80°C freezer for 2-4 hours
- Finally, store in liquid nitrogen vapor phase
These interrupted methods provide greater control over ice nucleation and crystal growth, potentially increasing post-thaw viability by 5-15% compared to standard single-rate protocols for challenging MSC sources [60].
Ice Nucleation Control Strategies
Controlled ice nucleation represents a critical yet often overlooked parameter in cryopreservation protocol optimization. Uncontrolled spontaneous nucleation introduces significant sample-to-sample variability and can compromise reproducibility in clinical manufacturing.
The Role of Ice Nucleation in Cryopreservation Quality
During slow freezing, the extracellular solution freezes first, creating a vapor pressure deficit that draws water out of cells. The temperature at which ice nucleation occurs directly impacts this process—lower nucleation temperatures result in greater supercooling and potentially more intracellular ice formation upon eventual nucleation [10]. Controlling this parameter ensures consistent freezing conditions across all samples, which is particularly crucial when scaling up processes for clinical manufacturing.
Industry surveys indicate that 87% of cell therapy developers now use controlled-rate freezing rather than passive freezing methods, reflecting recognition of the importance of parameter control for consistent product quality [38]. Those using passive freezing (13%) predominantly have products in earlier clinical stages, suggesting a transition to controlled freezing as therapies advance toward commercialization [38].
Practical Methods for Ice Nucleation Control
Manual Seeding Technique:
- Cool samples to -5°C to -10°C (slightly below the freezing point of the solution)
- Briefly touch the container with pre-cooled forceps or a cotton-tipped swab dipped in liquid nitrogen at the liquid-air interface
- Observe for ice formation, which should spread throughout the solution
- Continue controlled cooling at the prescribed rate
Automated Nucleation Systems:
- Newer controlled-rate freezers incorporate automated nucleation features
- Utilize a brief pulse of cold nitrogen vapor to initiate ice formation
- Provide superior reproducibility and documentation for GMP compliance
The optimal nucleation temperature typically falls between -5°C and -8°C for most MSC cryopreservation formulations, though this should be verified for specific CPA combinations [10]. Incorporating controlled nucleation can reduce post-thaw viability variation by up to 30%, significantly improving process robustness [38].
Advanced Research and Preconditioning Strategies
Cell Cycle Synchronization to Mitigate Cryo-Injury
Recent research has revealed that S phase MSCs are exceptionally sensitive to cryo-injury, demonstrating heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function [6]. This discovery has led to the development of novel preconditioning strategies that synchronize cells in less vulnerable cell cycle phases prior to freezing.
Serum Starvation Protocol for G0/G1 Synchronization:
- Culture MSCs to 70-80% confluence in standard complete medium
- Wash cells twice with phosphate-buffered saline (PBS) to remove serum residues
- Replace with serum-free medium for 24-48 hours
- Verify cell cycle distribution via flow cytometry (target: >70% in G0/G1)
- Proceed with standard trypsinization and cryopreservation protocols
This approach has demonstrated remarkable efficacy in preserving viability, clonal growth, and T cell suppression function at pre-cryopreservation levels, effectively eliminating the dysfunction typically associated with cryopreserved MSCs [6]. The mechanism involves preventing apoptosis induced by double-stranded breaks in labile replicating DNA that form during cryopreservation and thawing processes.
Alternative Strategy: Hydrogel Microencapsulation
Hydrogel microencapsulation technology presents an alternative approach to cryoprotection by creating a protective physical barrier around cells. Research demonstrates that alginate-based microcapsules enable effective cryopreservation of MSCs with as little as 2.5% DMSO while sustaining cell viability above the 70% clinical threshold [61].
The hydrogel matrix functions by:
- Providing a 3D environment that protects against mechanical ice damage
- Facilitating nutrient and gas exchange while shielding cells
- Potentially restricting ice crystal growth through physical confinement
- Enabling long-term cryopreservation without compromising viability
This approach is particularly valuable for clinical applications where DMSO toxicity presents significant concerns, including nausea, vomiting, arrhythmias, and neurotoxicity in patients [61].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful optimization of MSC cryopreservation requires access to specialized reagents and equipment. The following table details essential materials for implementing the protocols discussed in this guide.
Table 3: Essential Research Reagents and Materials for MSC Cryopreservation Optimization
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Programmable Controlled-Rate Freezer | Precisely controls cooling rate; essential for protocol optimization | Temperature range: +40°C to -180°C; cooling rate: 0.1°C to -50°C/min |
| DMSO (Cell Culture Grade) | Penetrating cryoprotectant; disrupts ice crystal formation | Sterile-filtered, tissue culture tested; typically used at 5-10% (v/v) |
| Serum-Free Medium | Cell cycle synchronization via serum starvation | With essential amino acids, glucose, and salts; without growth factors |
| Alginate Hydrogel System | Microencapsulation for reduced CPA requirements | High G-content alginate (>60%); sterile calcium chloride solution |
| Ice Nucleation Device | Controlled initiation of ice formation | Automated seeder or manual tools (pre-cooled forceps) |
| Viability/Cell Cycle Assays | Post-thaw assessment of recovery and function | Flow cytometry reagents for apoptosis and cell cycle analysis |
Optimizing cooling rates and ice nucleation control for different MSC sources requires a systematic approach that addresses both the physical challenges of ice formation and the biological vulnerabilities of these therapeutic cells. The strategies outlined in this guide—from fundamental cooling rate adjustments to advanced preconditioning methods—provide a roadmap for significantly enhancing post-thaw recovery and functionality.
Future directions in MSC cryopreservation research will likely focus on several emerging areas. Computational modeling and AI-driven protocol optimization are showing promise in predicting optimal cooling parameters for specific cell types and donor characteristics [62] [59]. The development of DMSO-free cryopreservation formulations continues to advance, with hydrogel encapsulation and nanoparticle-based cryoprotectants offering promising alternatives [61] [59]. Additionally, the integration of digital twin technology for cryopreservation process design may accelerate optimization while conserving resources [62].
As the field progresses toward increasingly personalized cell therapies, the ability to reliably preserve functional MSCs will remain fundamental to translating laboratory research into clinical practice. Through careful attention to the principles outlined in this guide, researchers can contribute to the development of more robust and effective cryopreservation protocols that maximize the therapeutic potential of these remarkable cells.
The cryopreservation of Mesenchymal Stem Cells (MSCs) is a critical step in the cell therapy pipeline, enabling the creation of cell banks, facilitating transport, and allowing for off-the-shelf availability of therapeutic products [10] [55]. However, the process of freezing and thawing inflicts significant stress and damage on cells, collectively known as cryo-injury. Within the broader context of cryo-injury mechanisms, two primary damaging events occur: physical damage from ice crystal formation and osmotic stress during water movement, and biochemical damage from the generation of reactive oxygen species (ROS) [27] [23]. These insults can lead to reduced post-thaw viability, impaired metabolic and adhesion potential, altered differentiation capacity, and induction of apoptosis [23] [55]. To mitigate these damages, the strategic incorporation of additives into cryoprotective media is essential. This whitepaper provides an in-depth technical guide on two key classes of these additives: antioxidants, which combat oxidative stress, and macromolecules, which primarily address physical and osmotic stress. Understanding their protective mechanisms is vital for developing optimized cryopreservation protocols that maintain the critical quality attributes of MSCs for research and clinical applications.
Understanding Cryo-injury in MSCs
Physical and Osmotic Mechanisms
The physical and osmotic damage during cryopreservation stems from the phase change of water. During slow freezing, extracellular ice formation increases the solute concentration in the unfrozen fraction, creating a hypertonic environment. This draws water out of the cell, leading to detrimental cell shrinkage and dehydration [27] [12]. If the cooling rate is too rapid, intracellular water does not have time to exit and instead freezes within the cell. The resulting intracellular ice crystals are mechanically destructive, physically disrupting organelles and the plasma membrane [12] [10]. The selection of an appropriate cooling rate is therefore a balancing act designed to minimize both excessive dehydration and intracellular ice formation.
Oxidative Stress Mechanisms
A less visible but equally damaging aspect of cryo-injury is oxidative stress. The freeze-thaw process disrupts cellular metabolism and electron transport chains, particularly in mitochondria, leading to a burst of reactive oxygen species (ROS) including superoxide (O₂•⁻) and hydrogen peroxide (H₂O₂) [23]. In the presence of metal ions like iron, these can be converted via the Fenton reaction into the highly damaging hydroxyl radical (•OH) [23]. Supraphysiological levels of ROS inflict damage on all major cellular components: they induce lipid peroxidation of cell membranes, oxidize amino acids leading to protein fragmentation and loss of function, and cause DNA damage [23]. This oxidative assault can trigger cascades of cellular dysfunction, including loss of membrane integrity, reduced metabolic activity, and activation of apoptotic pathways, ultimately compromising cell survival and function post-thaw [23] [55].
Table 1: Key Reactive Oxygen Species Involved in Cryo-injury
| ROS Species | Chemical Symbol | Key Characteristics | Primary Detoxification System |
|---|---|---|---|
| Superoxide | O₂•⁻ | Charged, poorly membrane-permeable, precursor to other ROS | Superoxide Dismutase (SOD) |
| Hydrogen Peroxide | H₂O₂ | Membrane-permeable, relatively stable, signaling molecule | Catalase, Glutathione Peroxidase |
| Hydroxyl Radical | •OH | Extremely reactive, highly damaging to all biomolecules | None (no known enzymatic pathway) |
The diagram below illustrates the interconnected pathways of cryo-injury and the points where protective additives intervene.
Antioxidant Additives: Combating Oxidative Stress
Mechanisms of Action
Antioxidants function as a cellular defense network by neutralizing ROS and preventing the propagation of oxidative chain reactions. Their mechanisms are diverse and often synergistic. A primary mode of action is direct scavenging of free radicals, where antioxidants donate an electron to stabilize highly reactive ROS like •OH and O₂•⁻, thereby terminating their damaging activity [23]. Some antioxidants, such as catalase, are enzymes that catalytically convert specific ROS into less harmful substances; catalase, for instance, decomposes H₂O₂ into water and oxygen [23]. Another crucial mechanism involves the chelation of transition metal ions like iron, which are potent catalysts for the generation of •OH via the Fenton reaction. By sequestering these metals, antioxidants prevent the initiation of this highly damaging pathway [23].
Key Antioxidants and Experimental Evidence
Extensive research has investigated both enzymatic and non-enzymatic antioxidants for cryopreservation.
- Ascorbic Acid (Vitamin C): This water-soluble vitamin is a potent direct scavenger of various ROS. In cryopreserved sperm models, the addition of ascorbic acid demonstrated a significant reduction in ROS levels and DNA damage, while improving cell viability and mitochondrial membrane potential [23]. Its efficacy, however, can be concentration-dependent and variable across cell types.
- Catalase: This enzyme provides highly efficient and specific detoxification of H₂O₂. Studies have shown its addition to cryopreservation media leads to reduced ROS and decreased levels of apoptotic and necrotic cells [23]. Its large molecular size may limit its access to intracellular compartments.
- Antifreeze Proteins (AFPs): While known for their ice-binding properties, AFPs also exhibit antioxidant capabilities. Research on vitrified oocytes showed that AFP supplementation resulted in lower ROS levels, reduced DNA damage (measured by γH2AX-positive cells), and improved developmental outcomes such as blastocyst rate [23].
- Coenzyme Q10 (CoQ10): This lipid-soluble antioxidant is integral to the mitochondrial electron transport chain and can effectively protect membranes from lipid peroxidation. In sperm cryopreservation, CoQ10 improved viability, membrane integrity, and reduced DNA fragmentation [23].
Table 2: Efficacy of Selected Antioxidants in Cryopreservation Models
| Antioxidant | Cell Type Studied | Reported Beneficial Effects | Potential Limitations |
|---|---|---|---|
| Ascorbic Acid | Sperm | ↓ ROS, ↑ Viability, ↓ DNA Damage [23] | Effects on motility inconsistent [23] |
| Catalase | Sperm | ↓ ROS, ↑ Viability & Motility, ↓ Apoptosis [23] | Large size, no effect on oocyte survival in one study [23] |
| Antifreeze Proteins (AFP) | Oocytes | ↓ ROS, ↑ Viability & Blastocyst Rate, ↓ DNA Damage [23] | Cost, potential immunogenicity |
| Coenzyme Q10 | Sperm | ↑ Viability & Motility, ↓ Lipid Peroxidation & DNA Fragmentation [23] | Lipophilicity, solubility challenges |
Detailed Experimental Protocol: Assessing Antioxidant Efficacy in MSC Cryopreservation
The following protocol outlines a standard procedure for evaluating the effect of an antioxidant, such as Ascorbic Acid, on the post-thaw quality of MSCs.
Objective: To determine the impact of Ascorbic Acid supplementation in cryopreservation media on the viability, metabolic activity, and oxidative stress levels of human Bone Marrow-MSCs (hBM-MSCs) post-thaw.
Materials:
- Passage 4 hBM-MSCs
- Standard growth medium (e.g., DMEM with 10% FBS)
- Freezing vehicle (e.g., Autologous plasma or FBS)
- Cryoprotectant (e.g., 10% DMSO)
- Antioxidant: L-Ascorbic Acid (prepare a stock solution in PBS or media, filter sterilize)
- Control cryomedium: Freezing vehicle + 10% DMSO
- Test cryomedium: Freezing vehicle + 10% DMSO + Ascorbic Acid (e.g., 50-200 µM)
- Cryogenic vials, Mr. Frosty or controlled-rate freezer, water bath, centrifuge.
Methodology:
- Cell Preparation and Cryopreservation:
- Harvest P4 hBM-MSCs at ~80% confluence using standard trypsinization.
- Centrifuge the cell suspension and resuspend the pellet in the pre-chilled control or test cryomedium at a density of 1 x 10⁶ cells/mL.
- Aliquot 1 mL of the cell suspension into cryogenic vials.
- Place vials in a Mr. Frosty freezing container pre-cooled to 4°C, and transfer immediately to a -80°C freezer for 24 hours to achieve a cooling rate of approximately -1°C/min.
- After 24 hours, swiftly transfer the vials to a liquid nitrogen tank for long-term storage (minimum 1 week).
Thawing and Post-Thaw Analysis:
- Rapidly thaw the vials by agitation in a 37°C water bath for 1-2 minutes.
- Gently transfer the cell suspension to a tube containing 9 mL of pre-warmed growth medium to dilute the DMSO.
- Centrifuge at 300-400 x g for 5 minutes to pellet cells. Discard the supernatant.
- Resuspend the cell pellet in fresh growth medium and proceed with analyses.
Key Assays and Time Points:
- Viability and Apoptosis (0h, 4h, 24h post-thaw): Use Annexin V/PI staining and flow cytometry to distinguish live, early apoptotic, and necrotic populations [55].
- Metabolic Activity (4h, 24h post-thaw): Assess using an AlamarBlue or MTT assay. Note that metabolic activity typically remains depressed at 24h compared to fresh cells, even as viability recovers [55].
- Intracellular ROS (0h, 2h post-thaw): Measure using fluorescent probes like H2DCFDA for general ROS or DHE for superoxide, followed by flow cytometry or fluorescence microscopy [23].
- Functional Assays (After 24h recovery): Perform CFU-F assays to assess clonogenic potential and initiate trilineage differentiation assays (osteogenic, adipogenic, chondrogenic) to determine if differentiation capacity is retained [63] [55].
Data Analysis: Compare all endpoints between the control (no antioxidant) and test (with antioxidant) groups using fresh cells as a baseline. Statistical significance is typically determined using a one-way or two-way ANOVA with post-hoc tests (p < 0.05).
Macromolecular Additives: Mitigating Physical and Osmotic Damage
Mechanisms of Action
Macromolecules, typically high molecular weight polymers and sugars, are non-penetrating cryoprotective agents that exert their protective effects primarily in the extracellular space. Their action is fundamentally rooted in colligative properties and specific biophysical interactions. A key mechanism is the reduction of freezing point and inhibition of ice crystal growth by increasing the total solute concentration in the extracellular solution. This reduces the amount of freezable water and minimizes the mechanical damage caused by large, sharp ice crystals [27] [12]. Many macromolecules, particularly disaccharides like sucrose and trehalose, promote vitrification—the formation of a non-crystalline, glassy state upon cooling. This glassy matrix immobilizes the cells and prevents the destructive process of ice crystallization altogether [12] [64]. Furthermore, the "water replacement hypothesis" proposes that these sugars can hydrogen-bond to polar head groups of membrane phospholipids, substituting for water molecules that are removed during dehydration and thereby stabilizing the membrane's structural integrity [64].
Key Macromolecules and Experimental Evidence
- Sucrose and Trehalose: These disaccharides are among the most widely used non-penetrating cryoprotectants. They function as osmotic buffers, counteracting the excessive cell shrinkage caused by penetrating agents like DMSO. Their ability to form a stable glassy state and stabilize membranes is well-documented [27] [12]. Trehalose, found in desiccation-tolerant organisms, is particularly effective due to its high glass transition temperature and unique stability [12].
- Hydroxyethyl Starch (HES): A high molecular weight polymer, HES is a common additive in cryopreservation solutions. It remains exclusively extracellular and increases the solution viscosity, which can moderate ice crystal growth and reduce the rate of water efflux during freezing, thus minimizing osmotic shock [27].
- Polyvinylpyrrolidone (PVP) and Ficoll: These synthetic polymers are used as exocellular cryoprotectors. They contribute significantly to the vitrification tendency of the solution and help to maintain osmotic balance, thereby protecting the cell from the damaging effects of concentrated electrolytes [27].
- Serum Albumin and Polyethylene Glycol (PEG): Albumin is a natural protein component of many cryomedia, thought to coat cells and provide membrane stabilization. PEG, with its flexible polymer chains, can also interact with cell surfaces and has been reported to protect against freeze-thaw-induced ROS generation [27] [23].
Detailed Experimental Protocol: Optimizing a Macromolecule-Enhanced Cryomedium
This protocol details the formulation and testing of a cryoprotective medium combining a penetrating agent (DMSO) with non-penetrating macromolecules (Sucrose and HES) for MSCs.
Objective: To develop and evaluate a vitrification-inspired slow-freezing medium that minimizes DMSO concentration while maintaining high post-thaw MSC recovery and function.
Materials:
- Base solution (e.g., Plasma, or a defined electrolyte solution)
- Dimethyl Sulfoxide (DMSO)
- Macromolecules: Sucrose, Hydroxyethyl Starch (HES, e.g., MW 200,000)
- Control: 10% DMSO in Base Solution.
- Test Formulations:
- F1: 5% DMSO + 150mM Sucrose in Base Solution.
- F2: 5% DMSO + 150mM Sucrose + 5% HES in Base Solution.
Methodology:
- Medium Preparation: Prepare the cryomedia formulations fresh or as frozen stocks. Ensure all components are fully dissolved. Osmolality should be measured and recorded for each formulation.
- Cell Preparation and Freezing:
- Harvest and count MSCs as described in Section 3.3.
- Centrifuge and resuspend the cell pellet in the different test cryomedia and the control medium at a density of 1 x 10⁶ cells/mL.
- Follow the same freezing procedure using a Mr. Frosty or controlled-rate freezer as in Section 3.3.
- Thawing and Analysis:
- Thaw cells rapidly in a 37°C water bath.
- Dilute the thawed suspension 1:10 with pre-warmed culture medium. Note: For media containing high molecular weight polymers like HES, a single centrifugation step may be insufficient for complete removal. Consider using a density gradient or assessing the necessity of removal based on downstream assays.
- Centrifuge and resuspend for analysis.
- Key Assays:
- Post-Thaw Recovery: Calculate the percentage of viable cells recovered relative to the number frozen.
% Recovery = (Viable Cell Count Post-Thaw / Total Cells Frozen) * 100. - Membrane Integrity: Use a LIVE/DEAD (e.g., Calcein-AM/EthD-1) viability/cytotoxicity kit for fluorescence microscopy or flow cytometry.
- Adhesion Potential: Plate a known number of thawed cells and quantify the percentage that adhere and spread after 4-6 hours. This is a sensitive indicator of cryo-injury [55].
- Apoptosis Assay: Perform Annexin V/PI staining at 4h and 24h post-thaw to assess delayed-onset apoptosis.
- Clonogenic Assay (CFU-F): Plate a low density of cells (e.g., 300,000 cells in a 6-well plate) and culture for 14 days. Fix, stain with crystal violet, and count colonies (>50 cells) to assess stem cell functionality [63].
- Post-Thaw Recovery: Calculate the percentage of viable cells recovered relative to the number frozen.
The following diagram summarizes the workflow for this experiment, from formulation to analysis.
The Scientist's Toolkit: Research Reagent Solutions
The following table catalogs essential reagents for investigating the protective roles of antioxidants and macromolecules in MSC cryopreservation.
Table 3: Essential Reagents for Investigating Additives in MSC Cryopreservation
| Reagent / Kit | Function / Application | Technical Notes |
|---|---|---|
| L-Ascorbic Acid | Water-soluble antioxidant; scavenges various ROS. | Use stable, cell-culture grade forms. Test in 50-200 µM range; protect from light. |
| Catalase (from bovine liver) | Enzymatic antioxidant; specifically decomposes H₂O₂. | Large molecule (≈240 kDa). Add directly to cryomedium. Filter sterilize. |
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant; standard component of cryomedia. | Handle in a fume hood. Final concentration typically 5-10% (v/v). |
| Sucrose | Non-penetrating cryoprotectant; osmotic buffer, promotes vitrification. | Commonly used at 0.1-0.3 M. Contributes significantly to osmolality. |
| Trehalose (dihydrate) | Non-penetrating cryoprotectant; high glass transition temperature, membrane stabilizer. | Preferred over sucrose for some applications due to stability [12]. |
| Hydroxyethyl Starch (HES) | High molecular weight polymer; increases viscosity, modulates ice growth. | Does not penetrate cell. Can be used at 2-10% (w/v). May complicate post-thaw washing. |
| Annexin V-FITC / PI Apoptosis Kit | Distinguishes live, early apoptotic, and necrotic cell populations by flow cytometry. | Critical for assessing delayed-onset apoptosis at 4-24h post-thaw [55]. |
| H2DCFDA / DHE Fluorescent Probes | Cell-permeable dyes for detecting general ROS and superoxide, respectively. | Use with flow cytometry or fluorescence microscopy. Load cells post-thaw. |
| AlamarBlue / MTT Assay Kits | Measure cellular metabolic activity as a proxy for viability and health. | More sensitive than simple cell counting for detecting sublethal injury [55]. |
| Crystal Violet Solution | Stains cell nuclei for Colony-Forming Unit (CFU-F) fibroblast assays. | Allows quantification of clonogenic potential, a key MSC functionality test [63]. |
The strategic incorporation of antioxidants and macromolecules into cryopreservation media represents a sophisticated and multi-targeted approach to mitigating the complex mechanisms of cryo-injury in MSCs. While antioxidants directly counter the biochemical assault of oxidative stress, macromolecules address the physical and osmotic challenges of ice formation and solute concentration. The experimental evidence and protocols outlined in this whitepaper provide a foundation for researchers to systematically evaluate and optimize these additives. The ultimate goal is the development of robust, defined, and clinically compliant cryopreservation protocols that can be standardized across the industry. Future research directions should focus on a deeper mechanistic understanding using omics technologies (e.g., phosphoproteomics to identify key signaling pathways affected by cryo-injury and protection [65]), the exploration of synergistic combinations of additives, and the translation of these findings into xeno-free, serum-free, and potentially DMSO-free cryopreservation solutions for clinical-grade MSCs. By viewing cryopreservation not just as a storage method but as a critical unit operation in the cell manufacturing process, we can significantly enhance the quality, efficacy, and reliability of MSC-based therapies.
Pre-conditioning and 'Pre-licensing' of MSCs (e.g., with IFN-γ) Before Freezing
The clinical development of mesenchymal stromal cell (MSC)-based therapies has been significantly hindered by ineffective cryopreservation methods that result in substantial loss of post-thaw cell viability and function [6] [15]. Cryopreservation induces complex cryo-injury mechanisms that extend beyond immediate cell death, profoundly impairing critical therapeutic functions such as immunomodulation [7] [66]. Within this context, pre-conditioning or pre-licensing—the exposure of MSCs to bioactive molecules prior to freezing—has emerged as a strategic approach to enhance cellular resilience and preserve functionality post-thaw.
This technique is particularly relevant given recent insights into fundamental cryo-injury mechanisms. Research has revealed that S-phase MSCs are exquisitely sensitive to cryoinjury, demonstrating heightened levels of delayed apoptosis post-thaw and reduced immunomodulatory function [6] [15]. While pre-licensing with cytokines like interferon-gamma (IFN-γ) represents a promising solution, it must be evaluated within a comprehensive understanding of cryo-injury pathways and their impact on MSC biology.
Fundamental Mechanisms of Cryo-injury in MSCs
Cell Cycle-Dependent Cryosensitivity
A pivotal recent discovery identified that cryoinjury is not uniform across the MSC population but demonstrates marked cell cycle dependence. MSCs in the S-phase (DNA synthesis phase) are particularly vulnerable, showing heightened levels of delayed apoptosis post-thaw [6]. The underlying mechanism involves double-stranded breaks in labile replicating DNA that form during the cryopreservation and thawing processes [6]. This specific vulnerability presents a targeted opportunity for intervention strategies aimed at synchronizing cells in less vulnerable cell cycle stages prior to freezing.
Functional Impairment of Immunomodulatory Capacity
Beyond immediate cell death, cryopreservation can impair the immunomodulatory functions that are central to MSC therapeutic efficacy. Several studies report that cryopreserved MSCs may exhibit reduced potency in immunosuppression assays [7] [66]. Specifically:
- Susceptibility to Immune Lysis: Cryopreserved MSCs show increased susceptibility to lysis by cytotoxic T lymphocytes (CTLs) in co-culture systems, while fresh MSCs maintain their immune-evasive properties [7].
- Impaired Immunosuppressive Pathways: The in vitro immunosuppressive performance of frozen and thawed MSCs may be different from their fresh counterparts, with some studies showing a reduced, though not abolished, performance specifically in indoleamine 2,3-dioxygenase (IDO)-dependent pathways [66].
Table 1: Key Mechanisms of Cryo-injury in MSCs
| Mechanism | Cellular Consequence | Functional Impact |
|---|---|---|
| S-Phase Sensitivity | DNA double-stranded breaks; Delayed apoptosis | Reduced viability; Impaired clonal growth |
| Altered Immunomodulation | Failed suppression of T-cell degranulation; IDO pathway impairment | Reduced immunosuppressive capacity |
| Membrane Damage | Intracellular ice crystal formation; Osmotic stress | Immediate cell death; Loss of viability |
Pre-licensing Strategies and Protocols
IFN-γ Pre-licensing
Interferon-gamma pre-licensing aims to "prime" MSCs to maintain their immunosuppressive functions immediately upon thawing, bypassing the recovery period typically needed for cytokine response.
Experimental Protocol
The standard protocol involves treating MSCs with IFN-γ for 24-48 hours prior to cryopreservation [7]:
- Culture Conditions: Maintain MSCs in standard culture medium (e.g., DMEM low glucose supplemented with 10% platelet lysate or FBS) [7].
- IFN-γ Treatment: Add IFN-γ at concentrations typically ranging from 10-50 ng/mL to the culture medium.
- Incubation Period: Treat cells for 24-48 hours before harvesting for cryopreservation.
- Cryopreservation: Harvest cells using standard detachment methods and freeze in appropriate cryoprotectant medium.
Mechanistic Basis
IFN-γ pre-licensing works through several key mechanisms:
- IDO Upregulation: Pre-licensed cryopreserved MSCs exhibit high levels of IDO mRNA and protein immediately upon thawing, enabling immediate immunosuppressive capability [7].
- CTL Protection: IDO activity is known to suppress CD8+ cytotoxic T lymphocytes, protecting pre-licensed MSCs from CTL-mediated lysis [7].
Figure 1: Dual Consequences of IFN-γ Pre-licensing: Enhanced immunosuppression through IDO versus potential immune clearance via increased MHC expression.
Cell Cycle Synchronization as an Alternative Strategy
As an alternative to cytokine pre-licensing, recent research has explored cell cycle synchronization to mitigate S-phase-specific cryosensitivity.
Serum Starvation Protocol
Growth factor deprivation through serum starvation can synchronize MSCs in G0/G1 phase [6]:
- Standard Culture: Expand MSCs under standard culture conditions until 70-80% confluency.
- Serum Reduction: Replace complete medium with low-serum (e.g., 0.5-1% FBS) or serum-free medium.
- Incubation Period: Maintain cells in low-serum conditions for 24-72 hours before harvesting for cryopreservation.
- Validation: Confirm cell cycle synchronization through flow cytometry analysis of DNA content.
Efficacy Assessment
This approach has demonstrated remarkable effectiveness in preserving post-thaw function. Treated MSCs maintained viability, clonal growth, and T cell suppression function at pre-cryopreservation levels, performing equivalently to cells primed with IFN-γ [6].
Comparative Analysis of Pre-conditioning Strategies
Table 2: Comparative Efficacy of MSC Pre-conditioning Strategies
| Pre-conditioning Method | Reported Efficacy | Advantages | Limitations |
|---|---|---|---|
| IFN-γ Pre-licensing | Enhanced post-thaw immunomodulatory function; Protection from CTL lysis [7] | Immediate IDO activity post-thaw; Well-characterized mechanism | Pleiotropic effects may have unintended consequences; Increased MHC expression [7] |
| Cell Cycle Synchronization (Serum Starvation) | Viability and function preserved at pre-freeze levels; Reduces S-phase apoptosis [6] | Avoids cytokine exposure; Targets fundamental cryoinjury mechanism | Potential stress from nutrient deprivation; Requires optimization for different MSC sources |
| Combined IFN-γ + TNF-α | Protection in hostile environments (e.g., type 2 diabetes conditions) [67] | Robust and durable effect; Reverses palmitate-induced dysfunction | Complex protocol; Limited in vivo validation in cryopreservation context |
Functional Outcomes and In Vivo Evidence
In Vitro Potency Assessment
Multiple studies have quantified the effects of pre-licensing on MSC immunosuppressive capacity:
- IDO Expression Dynamics: MSCs pre-licensed with IFN-γ for 48 hours showed comparable IDO content at 8 and 24 hours after thawing to fresh MSCs continuously exposed to IFN-γ, while 24-hour pre-licensed MSCs showed reduced IDO at early time points [7].
- Immunosuppression Assays: Pre-licensed cryopreserved MSCs avoided CTL-mediated lysis while suppressing PBMC proliferation equally as well as fresh MSCs [7].
In Vivo Therapeutic Efficacy
The translation of in vitro findings to in vivo models presents a more complex picture:
- Disease-Specific Outcomes: While cryopreserved MSCs are effective in disease models of colitis, allergic airway inflammation, and ischemia/reperfusion injury, they may be unsuitable for other conditions like osteogenesis imperfecta [7].
- Context-Dependent Efficacy: In a retinal ischemia/reperfusion model, IFN-γ pre-licensed cryopreserved MSCs lost effectiveness in vivo, rescuing fewer retinal ganglion cells than either fresh or unlicensed cryopreserved MSCs [7].
- Immune Recognition Concerns: The pleiotropic effects of IFN-γ, including dramatically increased surface expression of MHC-I and MHC-II molecules, may accelerate immune detection and clearance of MSCs in allogeneic settings [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for MSC Pre-licensing and Cryopreservation Research
| Reagent / Solution | Function / Application | Representative Examples |
|---|---|---|
| IFN-γ | Primary pre-licensing cytokine; induces immunomodulatory enzymes | Human IFN-γ, Mouse IFN-γ |
| Cryoprotective Agents | Protect cells from freezing damage; enable vitrification | DMSO, Ethylene Glycol, Trehalose, Sucrose [27] [10] |
| Serum-Free/Low-Serum Media | Cell cycle synchronization through growth factor deprivation | DMEM with 0.5-1% FBS; Commercial serum-free MSC media |
| Viability Assays | Post-thaw cell integrity and function assessment | Propidium iodide, Acridine Orange, NucleoCounter [68] [69] |
| Immunosuppression Assay Components | Functional validation of immunomodulatory capacity | PBMCs from human donors; T-cell proliferation assays (e.g., CFSE) |
Pre-conditioning and pre-licensing strategies represent promising approaches to mitigate cryo-injury in MSC therapeutics. The choice between cytokine pre-licensing and cell cycle synchronization should be guided by the specific therapeutic application, route of administration, and immune context in which the MSCs will be used. While IFN-γ pre-licensing enhances immediate post-thaw immunomodulatory function, concerns about increased immunogenicity warrant careful consideration. Cell cycle synchronization through serum starvation offers a compelling alternative by addressing a fundamental mechanism of cryo-injury—S-phase vulnerability—without potentially detrimental immunogenic effects.
Future optimization of pre-conditioning strategies will require disease-specific validation and careful assessment of both the beneficial and potentially adverse effects of pre-licensing on MSC function and persistence in vivo. As cryopreservation remains essential for the practical implementation of MSC therapies, refining these pre-conditioning approaches will be crucial for developing potent, clinically effective off-the-shelf cellular products.
In the rapidly advancing field of mesenchymal stromal/stem cell (MSC)-based therapies, cryopreservation serves as a pivotal technological bridge, enabling the transition from laboratory research to clinically viable "off-the-shelf" medicinal products. The process of freezing and thawing, however, exposes cells to multiple stressors that can induce cryogenic injury, potentially compromising their therapeutic efficacy [70]. Within the context of a broader thesis on cryo-injury mechanisms, this technical guide establishes that the damage incurred during cryopreservation is not merely a matter of cell death but extends to more subtle alterations in cell function, phenotype, and potency. These alterations can significantly impact the consistency, predictability, and ultimately, the clinical success of MSC products [10] [70]. Therefore, implementing a rigorous, multi-parametric quality control (QC) regimen is not optional but fundamental to ensuring that thawed MSCs retain the critical attributes required for their intended biological function.
The inherent vulnerability of MSCs to cryo-injury stems from both physical and biochemical mechanisms. Physically, the formation of intracellular ice crystals can mechanically disrupt membrane integrity and organelle structures [27]. Biochemically, the process can induce osmotic stress and generate reactive oxygen species, leading to DNA damage, particularly in the replication-sensitive S-phase of the cell cycle [15] [27]. Furthermore, the disruption of the actin cytoskeleton post-thaw can impair cellular adhesion and engraftment potential [70] [46]. A comprehensive QC strategy must therefore extend beyond simple viability checks to probe the functional integrity of the cells, providing assurance that the thawed product is truly representative of its pre-freeze therapeutic potential.
Core Post-Thaw Quality Control Assessments
A robust QC framework for thawed MSCs is built upon four interdependent pillars: viability, recovery, phenotypic identity, and functional potency. Each pillar provides distinct yet complementary information essential for a holistic assessment of cell quality.
Viability and Recovery
Viability and cell recovery are the most immediate and fundamental metrics, serving as the first indicator of cryopreservation success. These parameters are typically assessed immediately post-thaw (0-hour) and can be monitored over a subsequent period (e.g., 2-6 hours) to gauge short-term stability [71] [72].
- Viability Measurement: The goal is to distinguish between live and dead cells. The Trypan Blue exclusion method is widely used for a quick assessment, where non-viable cells with compromised membranes take up the dye [71] [73]. For a more nuanced analysis, flow cytometry with Annexin V/Propidium Iodide (PI) staining is recommended. This method differentiates between viable (Annexin V-/PI-), early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cell populations, providing deeper insight into the mode of cell death triggered by cryo-injury [71] [72]. Studies aiming for clinical translation should target viabilities above 80-95% post-thaw, with minimal apoptosis [51] [46] [72].
- Cell Recovery Calculation: This metric quantifies the proportion of live cells recovered after thawing relative to the number that was cryopreserved. It is a critical measure of process efficiency.
% Viable Cell Recovery = (Total Live Cell Count Post-Thaw / Total Cell Count Cryopreserved) × 100Optimal recovery rates can exceed 90% with optimized protocols, indicating minimal loss due to cryo-injury and processing [46].
Table 1: Representative Post-Thaw Viability and Recovery Data from Published Studies
| Cryopreservation Solution | Post-Thaw Viability (%) | Viable Cell Recovery (%) | Key Findings | Citation |
|---|---|---|---|---|
| PHD10 (10% DMSO) | ~90% (0h, Trypan Blue) | Comparable to pre-freeze | Stable viability and recovery up to 6 hours post-thaw. | [71] |
| DMSO-Free (SGI) | ~83% (Overall) | ~92.9% | Slightly lower viability but superior recovery compared to some DMSO solutions. | [46] |
| Optimized 10% DMSO | >95% (TUNEL assay) | High | Demonstrated that high viability retention is achievable, preserving in vivo therapeutic potency. | [51] |
| FBS + 10% DMSO | >80% (Trypan Blue) | Not specified | Consistent performance, with fibroblasts retaining phenotype over 3 months of storage. | [73] |
Phenotypic Identity
Confirming that cryopreservation has not altered the fundamental identity of MSCs is crucial. This is assessed by verifying the expression of standardized surface markers as defined by the International Society for Cellular Therapy (ISCT). Cells must remain plastic-adherent and demonstrate a characteristic immunophenotype: positive for CD73, CD90, and CD105 (≥95%), and negative for hematopoietic markers CD45, CD34, CD14, CD19, and HLA-DR (≤2% positive) [10]. Flow cytometric analysis performed on thawed cells confirms that the freezing process has not selected for a subpopulation or induced aberrant marker expression. Multiple studies report that cryopreserved MSCs consistently maintain their canonical surface marker profile post-thaw, comparable to their fresh counterparts [71] [51] [72].
Functional Potency
Potency is the definitive quality attribute, confirming that the cells are not merely alive and correctly identified but also functionally competent. Given the primary mechanisms of action of MSCs in immunomodulation and paracrine signaling, potency assays should be tailored to the intended clinical application.
- Immunomodulatory Potency: A cornerstone assay involves the suppression of activated immune cell proliferation. In a standard protocol, thawed MSCs are co-cultured with CD3/CD28-activated peripheral blood mononuclear cells (PBMCs) or T-cells. The suppression of PBMC proliferation is typically measured by flow cytometry after 3-5 days using dyes like CFSE or via 3H-thymidine incorporation [51] [72]. Multiple studies have demonstrated that when post-thaw viability is high, MSCs maintain their ability to suppress T-cell proliferation effectively, with no significant difference from fresh cells [51] [72].
- Paracrine and Secretory Function: The ability of MSCs to respond to inflammatory cues and secrete trophic factors is vital. A key assay involves stimulating thawed MSCs with IFN-γ and/or TNF-α and measuring the upregulation of Indoleamine 2,3-dioxygenase (IDO), a critical immunomodulatory enzyme. IDO activity can be quantified by measuring the concentration of kynurenine, its metabolic product, in the supernatant via spectrophotometry [51]. Research confirms that cryopreserved MSCs can retain the capacity to upregulate IDO and produce kynurenine at levels similar to fresh cells [51].
- In Vivo Functional Validation: The most stringent test of potency is performance in a disease-relevant animal model. For instance, in a murine model of retinal ischemia/reperfusion injury, cryopreserved MSCs administered directly after thawing were as effective as fresh MSCs in rescuing retinal ganglion cells, providing direct evidence of retained therapeutic potency in a complex biological environment [51]. Similarly, in a polymicrobial sepsis model, thawed MSCs performed equivalently to cultured cells in improving bacterial clearance and reducing systemic inflammation [72].
Table 2: Key Functional Potency Assays for Thawed MSCs
| Functional Attribute | Example Assay | Readout | Interpretation |
|---|---|---|---|
| Immunomodulation | Co-culture with CD3/CD28-activated PBMCs | Reduction in PBMC proliferation (CFSE dilution) | Confirms retained capacity to modulate adaptive immune responses. |
| Response to Inflammatory Cue | Stimulation with IFN-γ (e.g., 48h) | IDO enzyme activity (Kynurenine concentration in supernatant) | Validates intact sensing and response machinery to inflammatory microenvironment. |
| Monocyte/Macrophage Modulation | Co-culture with LPS-impaired monocytes | Phagocytic capacity (e.g., uptake of fluorescent E. coli) | Assesses ability to enhance innate immune function. |
| In Vivo Potency | Administration in disease model (e.g., ischemia, sepsis) | Disease-specific metrics (e.g., cell survival, pathogen clearance, cytokine levels) | The gold standard for validating therapeutic efficacy post-thaw. |
Detailed Experimental Protocols
Thawing and Initial Processing
The thawing process itself is a critical source of stress and must be carefully controlled.
- Rapid Thawing: Remove the vial from liquid nitrogen storage and immediately place it in a 37°C water bath with gentle agitation until only a small ice crystal remains (approximately 1.5-2 minutes) [71]. To mitigate contamination risk from the water bath, the use of closed-system dry thawing devices is recommended for clinical-grade production [10].
- Dilution and CPA Removal: Immediately upon thawing, transfer the cell suspension to a pre-warmed tube containing a large volume (e.g., 10-fold) of culture medium or a specialized wash solution like Plasmalyte A with 5% Human Albumin (PLA/HA) [71]. This step rapidly dilutes the cytotoxic DMSO, minimizing its exposure time to the cells.
- Centrifugation: Centrifuge the cell mixture at a moderate force (e.g., 300-500 x g for 5-10 minutes) to pellet the cells.
- Resuspension and Counting: Carefully decant the supernatant and gently resuspend the cell pellet in fresh, complete culture medium. Perform a cell count and viability assessment using Trypan Blue or an automated cell counter.
In Vitro T-cell Suppression Assay
This protocol assesses the immunomodulatory potency of thawed MSCs [51] [72].
- PBMC Isolation: Isolate PBMCs from healthy donor buffy coats using density gradient centrifugation (e.g., Ficoll-Paque).
- CFSE Labeling: Label the PBMCs with a tracking dye such as Carboxyfluorescein succinimidyl ester (CFSE) according to the manufacturer's protocol.
- T-cell Activation: Activate the CFSE-labeled PBMCs using anti-CD3/CD28 activation dynabeads at a bead-to-cell ratio optimized for robust proliferation.
- Co-culture Setup: Plate thawed and washed MSCs in a 96-well plate and allow them to adhere for a few hours. Seed the activated PBMCs onto the MSC monolayer at defined ratios (e.g., MSC:PBMC ratios of 1:3, 1:6, 1:12). Include controls for unactivated PBMCs, activated PBMCs alone (maximum proliferation), and potentially fresh MSCs for comparison.
- Incubation and Analysis: Co-culture the cells for 3-5 days. Harvest the PBMCs and analyze CFSE fluorescence intensity by flow cytometry. The percentage of suppression is calculated by comparing the proliferation in co-culture wells to the maximum proliferation control.
% Suppression = [1 - (% Proliferated Cells in Co-culture / % Proliferated Cells in Activated Control)] × 100
IDO Activity Assay
This protocol measures the functional response of MSCs to inflammatory stimulation [51].
- Stimulation: Seed thawed MSCs at a defined density (e.g., 50,000 cells/cm²) and stimulate them with a cocktail of IFN-γ (e.g., 50 ng/mL) and/or TNF-α (e.g., 20 ng/mL) for 48-72 hours.
- Supernatant Collection: Collect the conditioned medium and centrifuge to remove any cellular debris.
- Kynurenine Measurement: Mix the supernatant with an equal volume of 2% (w/v) trichloroacetic acid and centrifuge. Then, transfer the supernatant to a fresh tube and mix with an equal volume of Ehrlich's reagent (p-dimethylaminobenzaldehyde in acetic acid).
- Quantification: Incubate the mixture for 10-15 minutes at room temperature and measure the absorbance at 490 nm. The kynurenine concentration is determined by comparison to a standard curve of known L-kynurenine concentrations.
The Scientist's Toolkit: Research Reagent Solutions
Successful post-thaw assessment relies on a suite of critical reagents and materials. The table below details essential components for a robust QC workflow.
Table 3: Essential Reagents and Materials for Post-Thaw QC
| Category/Item | Specific Examples | Function & Application Notes |
|---|---|---|
| Cryopreservation Media | PHD10 (Plasmalyte-A + 5% HA + 10% DMSO); Commercial Formulations (e.g., CryoStor CS10, CS5; NutriFreez) | Protect cells from cryo-injury during freeze-thaw. DMSO-free alternatives (e.g., SGI: Sucrose, Glycerol, Isoleucine) are emerging. [71] [46] |
| Thawing/Wash Media | Plasmalyte A with 5% Human Albumin (PLA/HA); Complete culture medium | Dilutes cytotoxic CPA post-thaw, provides osmotic support and nutrients during initial recovery. [71] |
| Viability & Apoptosis Assays | Trypan Blue; Annexin V/Propidium Iodide (PI) Apoptosis Detection Kit | Distinguish live, dead, apoptotic, and necrotic cell populations. Flow cytometry with Annexin V/PI is the gold standard. [71] [72] |
| Immunophenotyping | Fluorochrome-conjugated Antibodies against CD73, CD90, CD105, CD45, CD34, CD14, CD19, HLA-DR | Confirms MSC identity and purity post-thaw via flow cytometry, per ISCT guidelines. [10] [71] |
| Potency Assay Reagents | Anti-CD3/CD28 Dynabeads; CFSE Cell Division Tracker; Recombinant Human IFN-γ & TNF-α; L-Kynurenine Standard | Tools for T-cell suppression and IDO activity assays to validate immunomodulatory function. [51] [72] |
| Specialized Equipment | Controlled-Rate Freezer; 37°C Water Bath or Dry Thawing Device; Flow Cytometer; Spectrophotometer/Plate Reader | Enables standardized freezing, rapid thawing, and high-resolution analytical measurements. |
Data Interpretation and Mitigating Cryo-Injury
Interpreting post-thaw QC data requires an understanding of underlying cryo-injury mechanisms. A key finding is that cells in the S-phase of the cell cycle are exquisitely sensitive to cryopreservation, showing heightened levels of delayed apoptosis and DNA double-stranded breaks post-thaw [15]. This suggests that the cell cycle state at the time of freezing is a critical variable. Mitigation strategies include cell-cycle synchronization prior to freezing, such as via serum starvation to arrest cells in the G0/G1 phase, which has been shown to greatly reduce post-thaw dysfunction and preserve immunomodulatory function [15].
When QC data indicates suboptimal outcomes, the results guide troubleshooting. Consistently low viability may point to issues with the freezing rate, CPA toxicity, or thawing method. A study comparing revival methods found that a direct revival method (seeding without centrifugation) sometimes yielded a higher number of vials with optimal cell attachment, though an indirect method (with a centrifugation wash step) could result in higher expression of functional proteins like Ki67 and Collagen-1 after longer storage [73]. This highlights a trade-off between cell loss during processing and the benefits of CPA removal. Poor recovery might indicate osmotic shock during CPA addition or removal. Crucially, adequate viability without corresponding potency is a major red flag, suggesting that cells have survived but are functionally compromised, potentially due to cytoskeletal disruption or metabolic stunning that requires a recovery period in culture [70].
A comprehensive quality control strategy for thawed MSCs, integrating rigorous assessment of viability, recovery, phenotype, and—most critically—functional potency, is non-negotiable for the development of effective and reliable cell therapies. The presented checkpoints and protocols provide a framework for researchers to ensure that cryopreserved MSC products are not only viable but also therapeutically competent. As the understanding of cryo-injury mechanisms deepens, incorporating mitigation strategies such as cell-cycle synchronization and the development of novel, less toxic cryoprotectants will further enhance the quality of "off-the-shelf" MSC products, ultimately accelerating their successful translation to the clinic.
Functional Fitness: Assessing the Post-Thaw Potency and Clinical Readiness of Cryopreserved MSCs
The transition of mesenchymal stromal/stem cells (MSCs) from research tools to clinically viable "off-the-shelf" therapeutics hinges on effective cryopreservation. This technical review synthesizes current evidence on how cryopreservation impacts MSC viability and metabolic activity, framing these effects within the established mechanisms of cryoinjury. While cryopreservation introduces transient deficits in metabolic function and adhesion potential, a growing body of evidence indicates that key therapeutic properties can be preserved or recovered. The analysis provides detailed methodologies for assessing post-thaw cell quality and discusses emerging strategies to mitigate cryoinjury, offering researchers a foundation for developing robust, clinically applicable cryopreservation protocols.
The development of MSC-based therapies for acute conditions such as myocardial infarction, stroke, and acute lung injury necessitates readily available, "off-the-shelf" cell products [74] [51]. Cryopreservation enables biobanking, quality control, and immediate therapeutic access, yet concerns persist that freezing and thawing may compromise MSC viability and critical therapeutic functions. The core challenge lies in navigating the "lethality of an intermediate zone of temperature (-15 to -60 °C)" that cells must traverse during cooling and warming, where the physical and chemical stressors of cryopreservation manifest as cryoinjury [29]. This review quantitatively analyzes the impact of cryopreservation on MSC viability and metabolism, contextualizing findings within the mechanisms of cryoinjury to guide robust protocol development for clinical and research applications.
Quantitative Comparison of Viability and Metabolic Activity
Systematic comparisons reveal that cryopreservation induces immediate but often transient impairments in MSC viability and metabolic function. The degree of impairment and subsequent recovery are influenced by cryopreservation protocols, post-thaw handling, and the specific cell attribute measured.
Table 1: Post-Thaw Viability and Recovery of Cryopreserved MSCs
| Assessment Parameter | Immediate Post-Thaw (0-4h) | 24 Hours Post-Thaw | Key Findings | Reference |
|---|---|---|---|---|
| Viability (Trypan Blue) | Reduced | Recovered to near pre-freeze levels | Viability drops initially but recovers after 24h; delayed apoptosis manifests within first 4h. | [75] [76] |
| Apoptosis (Annexin V/PI) | Significantly increased | Significantly reduced | A 24h acclimation period post-thaw drastically reduces early and late apoptosis. | [77] |
| Metabolic Activity (XTT/Resazurin) | Significantly impaired | Remains lower than fresh cells | Metabolic activity is one of the most sensitive parameters, showing prolonged depression. | [75] [51] |
| Adhesion Potential | Impaired | Remains lower than fresh cells | Suggests cytoskeletal disruption, affecting engraftment potential post-infusion. | [75] |
| Clonogenic Capacity (CFU-F) | Not applicable | Reduced in 2 of 3 donor cell lines | Indicates variable impact on the proliferative potential of primitive progenitor populations. | [75] |
Table 2: Comparative Functional Potency of Fresh vs. Cryopreserved MSCs
| Functional Assay | Freshly Thawed MSCs | Cryopreserved MSCs (24h Acclimation) | Reference |
|---|---|---|---|
| Immunomodulatory Potency (T-cell Suppression) | Maintained, but potentially less potent | Significantly more potent; function regained | [77] |
| IDO Expression & Activity | Remains responsive to IFN-γ stimulation | Similar to fresh MSCs after 24h recovery | [51] [77] |
| In Vivo Efficacy (Preclinical Models) | ~97.7% of outcomes showed no significant difference vs. fresh | N/A | [74] |
| Proliferation Rate | Similar to fresh beyond 24h post-thaw | Similar to fresh beyond 24h post-thaw | [75] |
| Multipotent Differentiation | Maintained (osteogenic, chondrogenic) | Maintained (osteogenic, chondrogenic) | [77] |
A systematic review of pre-clinical models of inflammation found that the vast majority (97.7%) of in vivo efficacy outcomes showed no statistically significant difference between freshly cultured and cryopreserved MSCs [74]. This is a critical insight, suggesting that even with measurable in vitro deficits, cryopreserved MSCs can retain their core therapeutic function in complex biological systems.
Experimental Protocols for Assessing MSC Potency Post-Thaw
Time-Course Analysis of Viability and Metabolic Activity
Objective: To quantitatively track the recovery of cryopreserved MSCs in the first 24 hours post-thaw, a critical window for therapies intended for immediate administration.
Materials:
- Cryopreserved MSC vials (e.g., in 10% DMSO/FBS)
- Complete culture medium (e.g., α-MEM with 15% FBS)
- Trypan blue solution
- Annexin V/FITC and Propidium Iodide (PI) staining kit
- Metabolic assay kit (e.g., Resazurin-based Vybrant assay)
- Hemocytometer or automated cell counter
- Flow cytometer
- Fluorescence plate reader
Methodology:
- Thawing: Rapidly thaw MSC vials in a 37°C water bath for 1-2 minutes.
- Dilution & Washing: Transfer cell suspension to a pre-warmed complete medium (9:1 dilution), centrifuge at 200g for 5 minutes, and discard supernatant to remove DMSO.
- Resuspension & Plating: Resuspend cell pellet in fresh complete medium and perform an initial cell count and viability assessment (T0).
- Time-Course Incubation: Plate cells at a standardized density and harvest samples at T0, T2h, T4h, and T24h post-thaw for analysis.
- Viability & Apoptosis: Assess viability at each time point via Trypan blue exclusion. Quantify apoptosis and necrosis using Annexin V/PI staining followed by flow cytometry (viable: Annexin V-/PI-; early apoptotic: Annexin V+/PI-; late apoptotic/necrotic: Annexin V+/PI+).
- Metabolic Activity: Seed cells in a multi-well plate for the metabolic assay. At each time point, incubate with resazurin solution for 2-4 hours and measure fluorescence (Ex/Em ~563/587 nm). Normalize data to DNA content using a PicoGreen assay for accurate cell number correlation [75] [77].
Immunomodulatory Potency Assay
Objective: To determine the impact of cryopreservation on the immunosuppressive capacity of MSCs.
Materials:
- Peripheral Blood Mononuclear Cells (PBMCs) from human blood
- T-cell activator (e.g., CD3/CD28 dynabeads)
- IFN-γ
- Co-culture transwell plates
- CFSE cell proliferation dye or ^3H-thymidine
Methodology:
- MSC Preparation: Prepare three groups of MSCs from the same donor/passage: Freshly Cultured (FC), Freshly Thawed (FT), and Thawed + 24h Acclimation (TT).
- PBMC Activation: Isolate PBMCs and label with CFSE. Activate with CD3/CD28 dynabeads.
- Co-culture: Co-culture activated PBMCs with MSCs at varying ratios (e.g., 1:3, 1:6, 1:12 MSC:PBMC) in transwell systems for 3-5 days.
- Flow Cytometry Analysis: Harvest PBMCs and analyze CFSE dilution via flow cytometry to quantify T-cell proliferation suppression.
- ELISA: Collect supernatant to measure secretion of immunomodulatory factors like IFN-γ [51] [77].
Diagram 1: Immunomodulatory potency assay workflow.
Mechanisms of Cryoinjury and Impact on MSC Function
The quantitative deficits observed in cryopreserved MSCs are direct manifestations of well-characterized mechanisms of cryoinjury. Understanding these mechanisms is essential for developing targeted mitigation strategies.
Physical and Biochemical Pathways of Injury
Intracellular Ice Crystallization (IIF): At high cooling rates, intracellular water does not have sufficient time to efflux, leading to the formation of lethal ice crystals that mechanically disrupt organelles and the plasma membrane [26] [29]. This is a primary cause of immediate post-thaw cell death.
Solution Effects and Osmotic Imbalance: At slow cooling rates, extracellular ice formation increases solute concentration in the unfrozen fraction, creating a hypertonic environment. This leads to cell dehydration and osmotic shock, damaging membrane integrity and cellular proteins [76] [29].
Cryoprotectant (CPA) Toxicity: While DMSO is essential for preventing IIF, it is intrinsically cytotoxic at high concentrations and can induce cell differentiation or epigenetic modifications [77]. The toxicity is exacerbated during the addition and removal of CPAs if not performed with precise osmotic control.
Delayed Onset Apoptosis: Cells that survive the initial physical trauma of freezing may undergo programmed cell death hours later. This is linked to mitochondrial membrane disruption and the activation of caspase pathways, explaining the spike in apoptosis observed 2-4 hours post-thaw [75] [77].
DNA Damage in Replicating Cells: A recent mechanistic discovery identified that S-phase MSCs are exquisitely sensitive to cryoinjury. The cryopreservation process induces double-stranded breaks (DSBs) in the labile replicating DNA, leading to delayed apoptosis and loss of function post-thaw [6].
Functional Consequences on Metabolic and Adhesive Machinery
The mechanisms of cryoinjury directly target core cellular components, leading to the observed functional deficits:
- Metabolic Activity Depression: Osmotic stress and cold shock disrupt mitochondrial function, leading to reduced ATP production. This is measured as a decrease in resazurin reduction in metabolic assays [75] [51].
- Impaired Adhesion and Cytoskeletal Disruption: The actin cytoskeleton is particularly vulnerable to cryoinjury. Damage to this network impairs cell spreading and adhesion, critical first steps for engraftment after in vivo administration [75]. This also complicates in vitro assays that require cell attachment.
Diagram 2: Mechanisms of cryoinjury and functional deficits.
The Scientist's Toolkit: Key Reagents and Solutions
Table 3: Essential Research Reagents for MSC Cryopreservation Studies
| Reagent/Solution | Function/Application | Example Formulations | Critical Considerations |
|---|---|---|---|
| Permeating Cryoprotectant | Prevents intracellular ice formation by penetrating the cell. | DMSO (5-10%), Glycerol | Cytotoxic at high [ ] and upon prolonged exposure at RT; requires controlled addition/removal. |
| Non-Permeating Cryoprotectant | Protects extracellularly, mitigates osmotic shock. | Sucrose, Trehalose, Human Serum Albumin (HSA) | Often used in combination with DMSO to allow for lower DMSO concentrations. |
| Cryopreservation Media | Formulated solution for freezing cells. | 90% FBS + 10% DMSO; PLA/5% HA/10% DMSO (PHD10); Proprietary (e.g., CryoStor CS10) | Clinical transition requires moving from FBS to serum-free (e.g., HSA-based) formulations. |
| Recovery/Wash Medium | Dilutes and removes CPA post-thaw. | Complete culture medium (e.g., α-MEM + 15% FBS) | Must be pre-warmed; volume and centrifugation speed are critical to minimize osmotic damage. |
| Viability & Apoptosis Kits | Quantifies live, early/late apoptotic, and necrotic cells. | Trypan Blue, Annexin V/PI staining kit with flow cytometry | Annexin V/PI is more accurate than dye exclusion for detecting apoptosis post-thaw. |
| Metabolic Activity Assay | Measures cellular health and functional metabolism. | Resazurin (e.g., Vybrant), XTT | Results should be normalized to cell number (e.g., via DNA content) for accurate interpretation. |
Mitigation Strategies and Concluding Outlook
Emerging research points to several promising strategies to mitigate cryoinjury in MSCs. A primary approach is post-thaw acclimation. A 24-hour recovery period allows cells to repair the cytoskeleton, clear damaged components, and regain immunomodulatory potency, including the upregulation of angiogenic and anti-inflammatory genes [77]. Another innovative strategy is cell cycle synchronization. By arresting MSCs in the less vulnerable G0/G1 phase through serum starvation prior to freezing, researchers have successfully reduced post-thaw apoptosis and preserved immunomodulatory function by preventing DSBs in S-phase DNA [6]. Finally, optimizing cryopreservation formulations is crucial. Comparing clinical-ready solutions like PHD10 and NutriFreez demonstrates that solutions with 10% DMSO can maintain high viability and recovery up to 6 hours post-thaw, whereas lower DMSO concentrations (e.g., CS5) may show a decreasing trend in viability [76].
In conclusion, while cryopreservation imposes measurable, transient stresses on MSCs, leading to deficits in viability, metabolic activity, and adhesion, these challenges are not insurmountable. A comprehensive understanding of the mechanisms of cryoinjury—from intracellular ice formation to S-phase-specific DNA damage—informs the development of robust protocols. Through careful optimization of cryopreservation solutions, inclusion of a post-thaw acclimation period, and the adoption of novel pre-freeze conditioning strategies, the promise of functionally potent "off-the-shelf" MSC therapies can be fully realized.
The therapeutic success of mesenchymal stem cells (MSCs) in regenerative medicine is fundamentally rooted in their capacity to differentiate into multiple cell lineages, most notably osteocytes, adipocytes, and chondrocytes. This multilineage differentiation potential represents a critical quality attribute for cells destined to repair musculoskeletal tissues. However, when MSCs are subjected to cryopreservation—an essential process for cell banking, transport, and ensuring off-the-shelf availability—this functional capacity can be compromised through various mechanisms of cryo-injury. These injuries can induce molecular and physical stresses that impact cell viability, surface marker expression, and, crucially, the intricate signaling networks that govern differentiation. Understanding how cryopreservation affects the retention of osteogenic, adipogenic, and chondrogenic potential is therefore paramount for developing robust protocols that ensure clinical efficacy. This whitepaper synthesizes current research to provide a technical guide on assessing and preserving the trilineage differentiation capacity of MSCs post-cryopreservation, framed within the broader context of cryo-injury mechanisms.
Quantitative Data on Differentiation Potential Post-Cryopreservation
The following tables consolidate quantitative findings from key studies investigating the impact of cryopreservation and other variables on the multilineage differentiation potential of MSCs from various tissue sources.
Table 1: Effects of Cryopreservation on MSC Differentiation Potential
| Cell Type | Cryoprotectant/Medium | Differentiation Lineage | Quantitative Outcome Post-Cryopreservation | Citation |
|---|---|---|---|---|
| Rat Adipose-Derived MSCs (AD-MSCs) | Bambanker (Serum-free) | Adipogenic | Slight decrease in lipid droplet accumulation (9.57% ± 4.24 vs. 19.29% ± 6.51 in fresh cells, p=0.083). [78] | |
| Rat Adipose-Derived MSCs (AD-MSCs) | Bambanker (Serum-free) | Osteogenic | Calcium deposition largely preserved (18.94% ± 3.57 vs. 21.12% ± 5.01 in fresh cells). [78] | |
| Rat Adipose-Derived MSCs (AD-MSCs) | Bambanker (Serum-free) | Chondrogenic | Glycosaminoglycan accumulation maintained, comparable to fresh cells. [78] | |
| Human Knee MSCs | DMSO 10% or DMSO 10% + Sucrose 0.2M | Multilineage (Osteo, Adipo, Chondro) | Maintained differentiation potential after thawing while embedded in PRP-SF bioscaffold. [79] |
Table 2: Impact of Donor Age and Tissue Source on MSC Differentiation Potential (Fresh Cells)
| Cell Source | Donor Age | Osteogenic Potential | Chondrogenic Potential | Adipogenic Potential | Citation |
|---|---|---|---|---|---|
| Mouse AD-MSCs | N/A | High (Superior to iMEF, imBMSC, iCAL upon BMP9 stimulation). [80] | Information Not Specified | High upon BMP9 stimulation. [80] | |
| Equine BM-MSCs | Newborn | High ALP activity and proteoglycan content. [81] | High proteoglycan content. [81] | Information Not Specified | |
| Equine BM-MSCs | Geriatric (≥22 years) | Significant decline in ALP activity and calcium deposition. [81] | Significant decline in proteoglycan content. [81] | Information Not Specified | |
| Equine AT-MSCs | All Ages | Less affected by age for calcium deposition than BM-MSCs. [81] | Minimal chondrogenic potential across all age groups. [81] | Information Not Specified | |
| Human AD-MSC Subpopulation (CD31-/CD34+) | Adult | Information Not Specified | Information Not Specified | Highest adipogenic potential; highest expression of PPARγ and FABP4. [82] | |
| Wharton's Jelly MSCs | N/A | Poor baseline osteogenic commitment compared to BM-MSCs. [83] | Information Not Specified | Information Not Specified |
Experimental Protocols for Assessing Differentiation Potential
Standardized in vitro assays are crucial for quantifying the retention of differentiation potential after cryopreservation. Below are detailed protocols for inducing and analyzing trilineage differentiation.
Osteogenic Differentiation
- Induction Medium Composition: Dulbecco's Modified Eagle Medium (DMEM) with high glucose (4.5 g/L), supplemented with 10% fetal bovine serum (FBS), 0.1 μM dexamethasone, 10 mM β-glycerophosphate, and 50 μM ascorbate-2-phosphate. [84] Some protocols also include 10 nM vitamin D3. [84]
- Culture Method: Cells are seeded at a density of 1 x 10⁴ to 2 x 10⁴ cells/cm² and cultured in the induction medium for 21-28 days, with the medium changed every 2-3 days. [84]
- Assessment Methods:
- Cytochemical Staining: Fixed cultures are stained with 2% Alizarin Red S (pH 4.1-4.3) to detect calcium deposits. [79] [84]
- Quantitative Analysis: Alizarin Red staining can be extracted with 10% cetylpyridinium chloride and quantified by measuring absorbance at 540-562 nm. [81]
- Gene Expression Analysis: qRT-PCR for osteogenic markers such as Runx2, osteocalcin (OCN), osteonectin, and alkaline phosphatase (ALP). [84]
- Enzymatic Activity: ALP activity can be measured using p-nitrophenyl phosphate as a substrate and quantified at 405 nm. [81]
Adipogenic Differentiation
- Induction Medium Composition: DMEM with high glucose, 10% FBS, 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 100 μM indomethacin, and 1.745 μM (10 μg/mL) insulin. [85] [84]
- Culture Method: Cells are cultured to confluence and then stimulated with induction medium for 3-5 days, followed by a maintenance medium (DMEM-HG, 10% FBS, insulin) for 1-2 days. This cycle is typically repeated 2-3 times, with a final maintenance period for a total of 12-14 days. [85] [84]
- Assessment Methods:
- Cytochemical Staining: Fixed cells are stained with Oil Red O working solution to visualize intracellular lipid vacuoles. [79] [78]
- Quantitative Analysis: Oil Red O can be extracted with 100% isopropanol and quantified by measuring absorbance at 500-520 nm. [82]
- Gene Expression Analysis: qRT-PCR for adipogenic markers such as peroxisome proliferator-activated receptor gamma (PPARγ), fatty acid binding protein 4 (FABP4/aP2), and leptin. [85] [82]
Chondrogenic Differentiation
- Induction Medium Composition: A defined serum-free medium, typically DMEM with high glucose, supplemented with 1% ITS+ Premix (6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 ng/mL selenious acid, 1.25 mg/mL BSA, 5.35 μg/mL linoleic acid), 100 nM dexamethasone, 50 μg/mL ascorbate-2-phosphate, 40 μg/mL L-proline, and 10 ng/mL transforming growth factor-beta 1 (TGF-β1). [85] [84]
- Culture Method: The pellet culture system is the gold standard. 2.5 x 10⁵ cells are centrifuged in a conical polypropylene tube or a V-bottom 96-well plate to form a pellet, which is then cultured in suspension for 21-28 days. [85] [81]
- Assessment Methods:
- Histological Staining: Pellet sections are stained with Alcian Blue (at pH 2.5) or Safranin O to detect sulfated glycosaminoglycans (GAGs) in the cartilaginous matrix. [79] [81]
- Quantitative Analysis: GAG content can be quantified using a dimethylmethylene blue (DMMB) dye-binding assay. [81] Proteoglycan content in Safranin O-stained pellets can be quantified by measuring "Redness" in image analysis software. [81]
- Gene Expression Analysis: qRT-PCR for chondrogenic markers such as SOX9, collagen type II (COL2A1), and aggrecan (ACAN). [84]
Figure 1: Experimental workflow for evaluating the retention of multilineage differentiation potential in MSCs after cryopreservation. The process involves thawing cryopreserved cells and subjecting them to standardized lineage-specific induction protocols followed by quantitative and qualitative assessments.
Molecular Mechanisms of Differentiation and Impact of Cryo-Injury
The commitment of MSCs to a specific lineage is governed by complex and highly regulated transcriptional networks and signaling pathways. Cryopreservation-induced stress can disrupt these networks, leading to diminished differentiation capacity.
Key Signaling Pathways and Transcriptional Networks
- Osteogenesis: The Wnt/β-catenin and BMP/Smad pathways are primary drivers. BMP2/4/7 binding to receptors leads to Smad1/5/8 phosphorylation, which complexes with Smad4 and translocates to the nucleus to activate the master transcription factor RUNX2. RUNX2, in turn, upregulates osteogenic genes like osteocalcin (OCN) and osteopontin (OPN). The Hippo and JAK-STAT pathways also contribute. [86] [80] [83]
- Adipogenesis: This process is primarily regulated by the sequential activation of the C/EBP family and PPARγ. External cues induce expression of C/EBPβ and C/EBPδ, which then activate the expression of both C/EBPα and PPARγ. These two master regulators form a positive feedback loop and drive the expression of adipogenic genes like FABP4 and LPL. [85] [86]
- Chondrogenesis: The TGF-β/BMP signaling pathway is crucial, particularly TGF-β1 and TGF-β3. Signaling through Smad2/3 activates the expression of the master transcription factor SOX9. SOX9 is essential for the expression of key cartilage matrix components, including collagen type II (COL2A1) and aggrecan (ACAN). [84] [83]
Figure 2: Core transcriptional networks and signaling pathways governing MSC trilineage differentiation. Each lineage is driven by specific extracellular signals, intracellular mediators, and master transcription factors that activate lineage-specific gene programs. BMP = Bone Morphogenetic Protein; TGF-β = Transforming Growth Factor Beta; RUNX2 = Runt-Related Transcription Factor 2; PPARγ = Peroxisome Proliferator-Activated Receptor Gamma; SOX9 = SRY-Box Transcription Factor 9.
Mechanisms of Cryo-Injury on Differentiation Potential
Cryopreservation can impair differentiation efficiency through several interconnected mechanisms:
- Transcriptomic Alterations: Cryopreservation can alter the baseline gene expression profile of MSCs. Studies show that cryopreserved rat AD-MSCs exhibited reduced expression of the pluripotency marker REX1 and immunomodulatory markers TGFβ1 and IL-6, suggesting a broader impact on cellular function beyond mere viability. [78] A diminished baseline of key transcription factors could hinder the robust activation of differentiation pathways post-thaw.
- Disruption of Signaling Pathways: Ice crystal formation and osmotic stress during freezing and thawing can damage cell membranes and cytoskeletal structures. This physical damage can disrupt the integrity of membrane receptors and intracellular signaling complexes essential for sensing differentiation cues, such as those in the Wnt, TGF-β/BMP, and MAPK pathways. [80] [83]
- Energetic and Metabolic Stress: The cryopreservation process places significant metabolic stress on cells, depleting ATP reserves. As differentiation is an energy-intensive process requiring significant biosynthesis, cells recovering from cryopreservation may lack the metabolic capacity to efficiently execute the differentiation program. [78]
- Induction of Senescence: Cryo-injury can induce cellular senescence, a state of irreversible growth arrest. Senescent MSCs not only exhibit reduced proliferative capacity but also demonstrate markedly diminished differentiation potential, a phenomenon well-documented in replicative senescence. [85] [81]
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Reagents for MSC Differentiation Assays
| Reagent/Category | Specific Examples | Function in Differentiation |
|---|---|---|
| Basal Media | Dulbecco's Modified Eagle Medium (DMEM), high glucose (4.5 g/L) | Provides essential nutrients and energy for cell growth and matrix production. [85] |
| Induction Supplements | Dexamethasone, Ascorbate-2-phosphate, β-Glycerophosphate, IBMX, Indomethacin | Key inducers; Dexamethasone is a glucocorticoid used in all three lineages. Ascorbate is essential for collagen synthesis in osteo/chondrogenesis. β-Glycerophosphate provides phosphate for mineralization. IBMX and Indomethacin induce adipogenesis. [85] [84] |
| Growth Factors | Recombinant Human BMP-2, TGF-β1, TGF-β3, Insulin | Potent lineage-specific inducers. BMP-2 strongly promotes osteogenesis. TGF-β1/3 are crucial for chondrogenesis. Insulin is essential for adipogenesis. [85] [84] [83] |
| Serum/Supplements | Fetal Bovine Serum (FBS), ITS+ Premix (Insulin, Transferrin, Selenium) | Provides a broad spectrum of hormones, lipids, and attachment factors. ITS+ is used in defined, serum-free chondrogenic induction. [85] |
| Staining Kits | Alizarin Red S, Oil Red O, Alcian Blue | Histochemical dyes for detecting calcium deposits (Alizarin Red), lipid droplets (Oil Red O), and sulfated glycosaminoglycans (Alcian Blue). [79] [78] [84] |
| Cryoprotectants | Dimethyl Sulfoxide (DMSO), Sucrose | Penetrating (DMSO) and non-penetrating (sucrose) agents that protect cells from ice crystal formation during freezing. [79] |
The retention of multilineage differentiation potential following cryopreservation is a definitive benchmark for MSC quality and a critical determinant of their therapeutic utility. Evidence indicates that while cryopreservation can preserve basic differentiation capacity, it often leads to a quantifiable reduction in efficiency and alters key molecular signatures. The extent of this functional decline is influenced by a triad of factors: the cryopreservation protocol (including cryoprotectant choice and freezing rate), the tissue source of the MSCs (with AD-MSCs often showing robust osteo-adipogenic potential), and critical donor characteristics such as age. A deep understanding of the molecular mechanisms underlying differentiation, coupled with robust, quantitative assessment protocols, is indispensable for diagnosing cryo-injury and developing next-generation preservation strategies. Ensuring that MSCs retain their full functional repertoire post-thaw is not merely a technical challenge but a fundamental prerequisite for reliable and effective clinical outcomes in regenerative medicine.
Mesenchymal stem/stromal cells (MSCs) are multipotent cells characterized by their self-renewal capacity, differentiation potential, and potent immunomodulatory properties [87] [1]. These non-hematopoietic stem cells are defined by their adherence to plastic, specific surface marker expression (CD73, CD90, CD105), and lack of hematopoietic markers (CD34, CD45, CD14, CD19, HLA-DR) [10] [1]. Originally identified for their role in tissue repair, MSCs have emerged as critical regulators of immune responses, influencing both innate and adaptive immunity through direct cell contact and paracrine activity [87]. Their ability to suppress immune cell proliferation and modulate inflammatory environments makes them promising therapeutic agents for autoimmune diseases, graft-versus-host disease (GVHD), and other inflammatory disorders [87] [1].
The immunomodulatory functions of MSCs are not constitutive but are rather licensed by inflammatory signals within the microenvironment [87]. This review focuses on a central mechanism of MSC-mediated immunosuppression: the expression of indoleamine 2,3-dioxygenase (IDO) and its profound impact on immune cell proliferation and function, particularly in the context of cryopreservation challenges.
The IDO Pathway in MSC-Mediated Immunosuppression
IDO Enzyme Biology and Function
Indoleamine 2,3-dioxygenase (IDO) is a cytosolic, heme-containing enzyme that catalyzes the first and rate-limiting step in the degradation of the essential amino acid tryptophan along the kynurenine pathway [88]. This enzyme converts tryptophan to N-formylkynurenine, initiating a metabolic cascade that produces various immunoregulatory kynurenine metabolites [88] [89]. While two isoforms exist (IDO1 and IDO2), IDO1 is the primary enzyme responsible for immunomodulation in human MSCs and is inducible by inflammatory stimuli [88].
In human MSCs, IDO1 expression is typically low under basal conditions but is strongly induced by pro-inflammatory cytokines, particularly interferon-gamma (IFN-γ), often in synergy with tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) [88]. This induction represents a critical feedback mechanism whereby MSCs sense inflammation and respond with immunosuppressive activity to restore immunological homeostasis.
Mechanisms of Immune Suppression via IDO
MSC-derived IDO suppresses immune responses through two primary mechanisms:
- Tryptophan Depletion: By catabolizing local tryptophan, IDO creates a microenvironment deficient in this essential amino acid. Immune cells, particularly T lymphocytes, are exquisitely sensitive to tryptophan starvation, which activates metabolic checkpoints and inhibits cell cycle progression, leading to proliferative arrest and anergy [88] [89].
- Production of Kynurenines: The bioactive metabolites generated by tryptophan catabolism, collectively known as kynurenines, exert direct immunomodulatory effects. These metabolites can induce apoptosis of activated T cells and promote the differentiation and expansion of regulatory T cells (Tregs) [89]. The kynurenine pathway metabolite quinolinic acid can also exert neurotoxic effects under certain conditions [88].
Table 1: Key IDO-Generated Tryptophan Metabolites and Their Immunological Effects
| Metabolite | Immunological Function | Receptor/Mechanism |
|---|---|---|
| Kynurenine | Promotes Treg differentiation; induces T-cell anergy | Aryl hydrocarbon receptor (AHR) activation [89] |
| Quinolinic Acid | Neurotoxicity; T-cell apoptosis | NMDA receptor agonist [88] |
| 3-Hydroxyanthranilic Acid | Induces oxidative stress and apoptosis in neurons | Reactive radical species generation [88] |
The significance of this pathway is evident in physiological immune regulation, most notably in preventing maternal T-cell-mediated rejection of the fetal allograft during pregnancy, where placental cells express IDO to create an immunoprivileged environment [88].
Quantitative Analysis of IDO-Mediated Immune Suppression
The immunosuppressive capacity of MSCs via IDO can be quantified through various experimental metrics, including enzyme activity levels, changes in immune cell populations, and functional proliferation assays.
Table 2: Quantitative Metrics of IDO-Mediated Immunosuppression by MSCs
| Parameter Measured | Experimental Method | Typical Findings with MSC Coculture | Significance |
|---|---|---|---|
| IDO Enzyme Activity | HPLC measurement of tryptophan depletion/kynurenine production [88] | Up to 90% tryptophan depletion in culture medium [89] | Direct measure of functional IDO expression |
| T-cell Proliferation | CFSE dilution or ^3H-thymidine incorporation [87] | 50-80% inhibition of T-cell proliferation [87] | Functional readout of immunosuppressive potency |
| Treg Induction | Flow cytometry for CD4+CD25+FOXP3+ cells [87] [89] | 2-4 fold increase in Treg populations [89] | Measure of immune tolerance induction |
| Th17 Suppression | ELISA for IL-17A; intracellular cytokine staining [87] | Significant inhibition of IL-17A production [87] | Measure of anti-inflammatory effect on pro-inflammatory T-cells |
The efficacy of IDO-mediated immunosuppression varies depending on MSC tissue source. Comparative studies suggest that adipose tissue-derived MSCs (A-MSCs) may exert more potent immunomodulatory effects than bone marrow-derived MSCs (BM-MSCs), while umbilical cord-derived MSCs (UC-MSCs) demonstrate minimal risk of initiating an allogeneic immune response, making them attractive therapeutic candidates [87].
Experimental Protocols for Assessing IDO Function
Protocol 1: In Vitro T-cell Suppression Assay
Purpose: To quantify the functional capacity of MSCs to suppress T-cell proliferation via IDO activity.
Materials:
- MSCs (test and control groups)
- Peripheral blood mononuclear cells (PBMCs) from healthy donors
- Anti-CD3/CD28 activation beads or mitogens (e.g., PHA)
- Recombinant human IFN-γ
- IDO inhibitor (e.g., 1-methyl-tryptophan)
- CFSE cell proliferation dye
- Flow cytometer
Methodology:
- Seed MSCs in 96-well plates at varying ratios (e.g., 1:10 to 1:100 MSC:PBMC) and allow to adhere overnight.
- Pre-treat MSCs with 50-100 ng/mL IFN-γ for 24-48 hours to induce IDO expression. Include controls with 1-MT inhibitor (500 µM).
- Isolate PBMCs and label with CFSE (5 µM) according to manufacturer's protocol.
- Activate CFSE-labeled PBMCs with anti-CD3/CD28 beads (1 bead per cell) or PHA (5 µg/mL).
- Co-culture activated PBMCs with pre-treated MSCs for 4-5 days.
- Harvest cells and analyze CFSE dilution in CD3+ T-cells by flow cytometry to determine proliferation indices.
- Collect supernatant for kynurenine/tryptophan measurement by HPLC or ELISA to correlate with functional suppression.
Protocol 2: IDO Expression and Activity Analysis
Purpose: To directly measure IDO expression and enzymatic activity in MSCs.
Materials:
- MSC lysates and culture supernatants
- IFN-γ
- TRIzol reagent for RNA isolation
- qRT-PCR equipment and primers for IDO1
- Western blot equipment and anti-IDO1 antibody
- HPLC system or commercial kynurenine ELISA kit
Methodology:
- Treat MSCs with IFN-γ (50 ng/mL) for 6h (RNA), 24h (protein), and 48h (supernatant).
- Gene Expression: Extract total RNA, synthesize cDNA, and perform qRT-PCR using IDO1-specific primers. Normalize to housekeeping genes (GAPDH, β-actin).
- Protein Expression: Lyse cells, separate proteins by SDS-PAGE, transfer to membrane, and probe with anti-IDO1 antibody. Detect by chemiluminescence.
- Enzymatic Activity: Collect cell culture supernatant. Either:
- Analyze by HPLC to quantify kynurenine production (detection at 360 nm).
- Use commercial kynurenine ELISA kit per manufacturer's instructions.
- Normalize IDO activity to total cellular protein or cell number.
Impact of Cryopreservation on MSC Immunomodulatory Function
Cryopreservation is essential for the storage and distribution of MSC-based therapies but induces significant stress and damage to cells, potentially compromising their therapeutic efficacy, including immunomodulatory function [10]. The standard slow-freezing method, which cools cells at a controlled rate (approximately -1°C/min to -3°C/min) in the presence of cryoprotectants like dimethyl sulfoxide (DMSO), achieves approximately 70-80% cell survival but can impair critical cellular functions [10].
A fundamental mechanism of cryoinjury relevant to immunomodulation is the exquisite sensitivity of S-phase MSCs to freezing-induced damage. Cells actively replicating DNA during cryopreservation sustain heightened levels of delayed apoptosis post-thaw and demonstrate reduced immunomodulatory function, including impaired T-cell suppression [6]. This injury is mechanistically linked to double-stranded breaks in labile replicating DNA that form during the cryopreservation and thawing processes [6].
Table 3: Effects of Cryopreservation on MSC Immunomodulatory Properties
| Functional Attribute | Impact of Cryopreservation | Mitigation Strategy |
|---|---|---|
| Viability/Recovery | ~70-80% with slow freezing; delayed apoptosis in S-phase cells [10] [6] | Cell cycle synchronization at G0/G1 via serum starvation [6] |
| IDO Expression/Activity | Can be impaired due to cellular stress | Pre-cryopreservation priming with IFN-γ; post-thaw recovery culture [6] |
| T-cell Suppression | Significantly reduced in S-phase cells post-thaw [6] | G0/G1 cell cycle arrest prior to freezing preserves function [6] |
| Paracrine Signaling | Altered secretome profile; vesicle integrity may be compromised | Optimized freezing protocols; DMSO-free cryoprotectants [10] |
A promising strategy to mitigate this specific cryoinjury involves cell cycle synchronization prior to freezing. Growth factor deprivation (serum starvation) blocks cell cycle progression at the G0/G1 phase, greatly reducing post-thaw dysfunction by preventing apoptosis associated with replication-associated DNA damage [6]. This approach preserves viability, clonal growth, and T-cell suppression function at pre-cryopreservation levels, comparable to cells primed with IFN-γ [6].
Diagram 1: Cryoinjury mechanism and mitigation via cell cycle sync.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Investigating IDO-Mediated Immunomodulation
| Reagent/Category | Specific Examples | Research Function |
|---|---|---|
| Pro-Inflammatory Cytokines | Recombinant human IFN-γ, TNF-α, IL-1β [87] [88] | Licensing signal to induce IDO expression in MSCs |
| IDO Inhibitors | 1-Methyl-D/L-tryptophan (1-MT) [88] | Pharmacological inhibition to confirm IDO-specific mechanisms |
| Cell Proliferation Assays | CFSE, EdU, ^3H-Thymidine incorporation [6] | Quantitative measurement of immune cell proliferation suppression |
| Flow Cytometry Antibodies | Anti-CD3, CD4, CD8, CD25, FOXP3 [87] | Immunophenotyping of T-cell subsets and Treg identification |
| Molecular Analysis Tools | IDO1 qPCR primers, anti-IDO1 antibodies [88] | Assessment of IDO expression at gene and protein levels |
| Metabolite Detection | HPLC systems, Kynurenine ELISA [88] [89] | Direct quantification of IDO enzymatic activity |
| Cryopreservation Reagents | DMSO, Serum-free cryomedium [10] | Standard cell freezing and storage |
Signaling Pathways in IDO-Mediated Immunoregulation
The immunoregulatory effects of MSC-derived IDO extend beyond simple tryptophan depletion, involving complex signaling circuits that establish and maintain immune tolerance, particularly through the kynurenine-AHR axis.
Diagram 2: IDO-mediated immunosuppression signaling pathway.
Activation of the aryl hydrocarbon receptor (AHR) by kynurenine initiates a genetic program leading to the differentiation of naive T-cells into regulatory T-cells (Tregs) rather than inflammatory effectors [89]. Furthermore, this Kyn-AHR axis in dendritic cells drives them toward a tolerogenic phenotype, which further amplifies immunosuppression by producing anti-inflammatory cytokines like TGF-β and IL-10, and reinforcing the IDO-AHR feedback loop [89]. This cascade establishes "infectious tolerance," where tolerance spreads from one immune cell population to another, creating a durable immunosuppressive microenvironment [89].
The suppression of immune cell proliferation via IDO expression represents a cornerstone of MSC-mediated immunomodulation. This sophisticated mechanism, central to the therapeutic potential of MSCs, is critically vulnerable to cryopreservation-induced damage, particularly in replicating cells. Understanding the intersection between IDO biology and cryoinjury—and implementing strategies like cell cycle synchronization to mitigate it—is essential for advancing robust, effective, and clinically reliable MSC-based therapies. Future research must continue to bridge fundamental immunology with cryobiology to ensure that the immense therapeutic potential of MSCs is fully realized in clinical practice.
1. Introduction
The therapeutic application of Mesenchymal Stem Cells (MSCs) has traditionally centered on their capacity for differentiation and engraftment. However, a paradigm shift has occurred with the recognition that their primary mechanism of action is largely attributable to paracrine signaling—the secretion of a complex mixture of bioactive factors collectively known as the secretome [90] [1]. This secretome, comprising growth factors, cytokines, chemokines, and extracellular vesicles (EVs), empowers resident cells to regenerate damaged tissue, modulates immune responses, and enhances angiogenesis [90]. Within the specific context of MSC cryopreservation research, understanding the secretome is paramount. The core thesis is that cryo-injury from freezing and thawing processes can alter the composition and potency of this secreted factor cocktail, thereby compromising the therapeutic efficacy of MSCs post-thaw. This technical guide provides an in-depth analysis of secretome components and detailed methodologies for their analysis, framed by the critical need to assess and mitigate cryo-injury in regenerative medicine.
2. The MSC Secretome: Composition and Functional Significance
The MSC secretome is not a static entity; its profile is dynamically "personalized" according to microenvironmental cues, including the cellular stress imposed by cryopreservation [90]. A cryo-injured cell may exhibit a skewed secretory profile, potentially diminishing regenerative capacity and amplifying undesirable inflammatory responses.
- Soluble Factors: This category includes a wide array of proteins with distinct regenerative functions. Key groups include:
- Immunomodulatory Factors: Such as Prostaglandin E2 (PGE2), Indoleamine 2,3-dioxygenase (IDO), and Tumor Necrosis Factor-Stimulated Gene 6 (TSG-6), which regulate innate and adaptive immune responses [90].
- Trophic and Growth Factors: Including Vascular Endothelial Growth Factor (VEGF), Fibroblast Growth Factor (FGF), Hepatocyte Growth Factor (HGF), and Transforming Growth Factor-beta (TGF-β), which drive angiogenesis, cell proliferation, and matrix synthesis [90] [91].
- Extracellular Vesicles (EVs): These membrane-bound nanoparticles (exosomes, microvesicles) facilitate intercellular communication by transferring proteins, lipids, and nucleic acids (e.g., miRNA) to recipient cells, mediating many of the systemic effects of MSCs [90].
Table 1: Key Functional Groups within the MSC Secretome and Their Roles
| Functional Group | Key Factors | Primary Documented Actions | Relevance to Tissue Repair |
|---|---|---|---|
| Immunomodulation | PGE2, IDO, IL-1RA, TSG-6, IL-10 [90] | Suppresses T-cell proliferation; induces macrophage switch to anti-inflammatory M2 phenotype; inhibits neutrophil infiltration. | Resolves chronic inflammation, creates a pro-regenerative microenvironment. |
| Angiogenesis | VEGF, FGF, HGF, Angiopoietin [90] [91] | Promotes proliferation and migration of endothelial cells; stimulates formation of new blood vessels. | Restores oxygen and nutrient supply to ischemic or damaged tissues. |
| Cell Proliferation & Migration | FGF, VEGF, EGF, TGF-α [91] | Enhances mitogenic activity and motility of keratinocytes, fibroblasts, and other tissue-specific progenitors. | Accelerates re-epithelialization and granulation tissue formation in wound healing. |
| Anti-apoptosis & Matrix Remodeling | HGF, TGF-β, FGF, IGF-1 [90] | Inhibits programmed cell death; modulates fibroblast activity and collagen deposition. | Enhances cell survival in harsh microenvironments; improves quality of regenerated tissue. |
3. Methodologies for Secretome Production and Analysis
Standardized protocols are critical for generating reproducible and therapeutically relevant secretome preparations, especially for comparing the secretory profiles of fresh versus cryopreserved MSCs [92].
3.1. Experimental Workflow for Secretome Analysis The following diagram outlines a generalized workflow for the production, collection, and analysis of the MSC secretome.
3.2. Secretome Production and Collection Protocols
- Preconditioning Strategies: To enhance secretome potency, MSCs can be "preconditioned" prior to collection. This is particularly relevant for reversing damage associated with cryo-injury.
- Hypoxic Culture: Mimicking physiological oxygen conditions (1-10% O₂) upregulates HIF-1α, boosting secretion of pro-angiogenic factors like VEGF and angiopoietin [92].
- 3D Culture Systems: Culturing MSCs as spheroids or in hydrogels more closely mimics their native microenvironment, enhancing anti-inflammatory and tissue-regenerative properties compared to standard 2D monolayers [92].
- Biochemical Stimulation: Priming with inflammatory cytokines like IFN-γ and TNF-α can enhance the immunomodulatory profile of the secretome, increasing the yield of factors like PGE2, IDO, and IL-10 [92].
- Standardized Collection Protocol: The following steps are adapted from current methodologies to ensure a cell-free, well-characterized secretome [92]:
- Cell Culture: Grow MSCs to 70-80% confluence.
- Washing: Rinse cells thoroughly with phosphate-buffered saline (PBS) to remove residual serum proteins.
- Serum-Free Incubation: Incubate cells with a defined, serum-free medium for 24-48 hours. Critical Note: Using serum-free medium is essential to avoid contaminating the secretome with exogenous proteins from Fetal Bovine Serum (FBS) [92].
- Collection: Collect the conditioned medium (CM).
- Clarification: Centrifuge CM at 300 × g for 5 minutes to remove cells and debris.
- Sterile Filtration: Filter the supernatant through a 0.22 µm syringe filter to eliminate any remaining particles or microbes.
- Concentration & Storage: Concentrate the filtrate using centrifugal filter units (e.g., 3 kDa cutoff) and store at -80°C, preferably after lyophilization (freeze-drying) for long-term stability [92].
3.3. Analytical Techniques for Secretome Characterization
A multi-modal approach is required to fully characterize the complex secretome.
- Protein Quantification and Identification:
- Bicinchoninic Acid (BCA) Assay: A standard colorimetric method for determining the total protein concentration in the secretome preparation [93].
- Multiplex Immunoassays: Platforms like the Bio-Plex Pro Human Cytokine Panel allow for the simultaneous quantification of dozens of specific cytokines, chemokines, and growth factors from a small sample volume [94].
- Enzyme-Linked Immunosorbent Assay (ELISA): Used for absolute quantification of specific, high-interest targets (e.g., VEGF, IL-10) [90].
- Mass Spectrometry (MS): Provides an untargeted, high-throughput method for identifying and quantifying the entire proteomic profile of the secretome, enabling the discovery of novel biomarkers affected by cryo-injury [92].
- Extracellular Vesicle Characterization:
- Nanoparticle Tracking Analysis (NTA): Measures the size distribution and concentration of EVs in suspension.
- Transmission Electron Microscopy (TEM): Visualizes the morphology and ultrastructure of isolated EVs [93].
- Western Blot (WB): Confirms the presence of EV-specific marker proteins (e.g., CD63, CD81, TSG101) and the absence of negative markers (e.g., calnexin).
Table 2: Key Experimental Parameters for Secretome Production and Their Impact
| Parameter | Standard Condition | Optimization/Perturbation | Documented Effect on Secretome |
|---|---|---|---|
| Culture Format | 2D Monolayer | 3D Spheroid/Hydrogel [92] | Enhances anti-inflammatory and regenerative properties; better mimics physiology. |
| Oxygen Concentration | Normoxia (21%) | Hypoxia (1-10%) [92] | Upregulates HIF-1α, boosting angiogenic factors (VEGF, Angiotensin). |
| Biochemical Stimuli | Basal Medium | IFN-γ, TNF-α, H₂O₂ [92] | Increases immunomodulatory (PGE2, IDO) and pro-angiogenic factors. |
| Serum Presence | With FBS | Serum-free [92] | Critical. Prevents contamination with exogenous proteins for clean analysis. |
| Collection Time | 24-48 hours | Varies (e.g., 6h, 72h) | Influences factor concentration and profile; requires empirical optimization. |
4. The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their functions for conducting secretome analysis, with a focus on assessing post-thaw MSC function.
Table 3: Essential Reagents for Secretome and Paracrine Factor Research
| Reagent / Kit | Function / Application | Technical Notes |
|---|---|---|
| Defined Serum-Free Medium | Base medium for secretome collection. | Eliminates confounding FBS proteins; formulations like MEM Alpha are commonly used [93]. |
| Bicinchoninic Acid (BCA) Assay Kit | Colorimetric quantification of total protein concentration in conditioned medium. | Essential for normalizing sample loading in downstream assays [93]. |
| Multiplex Cytokine Array (e.g., Bio-Plex Pro) | Simultaneous quantification of multiple cytokines, chemokines, and growth factors. | Ideal for comprehensive secretory profiling with low sample volume [94]. |
| ELISA Kits (VEGF, IL-10, HGF, etc.) | High-sensitivity, absolute quantification of specific target factors. | Used for validating multiplex data or focusing on key analytes of interest. |
| 0.22 µm Syringe Filter | Sterile filtration of conditioned medium to remove cells and debris. | A critical step for preparing a cell-free secretome for analysis or therapy [92]. |
| Ultrafiltration Centrifugal Units (3 kDa MWCO) | Concentration and desalting of secretome proteins from conditioned medium. | Enables the analysis of low-abundance factors; used prior to proteomics or functional assays. |
| Genipin / Glutaraldehyde | Crosslinking agents for biomaterial encapsulation (e.g., chitosan microspheres). | Used in advanced delivery systems for sustained secretome release [93]. |
| Antibody Panels (CD73, CD90, CD105) | Flow cytometry validation of MSC phenotype post-thaw. | Confirms MSC identity and viability after cryopreservation, a prerequisite for secretome studies [1]. |
5. Advanced Delivery and Engineering Strategies
To overcome the short half-life and poor tissue penetration of the native secretome, advanced delivery systems are being developed.
- Magneto-Actuated Micromotors: As explored in cutting-edge research, magnetic chitosan microspheres can be loaded with the secretome (CSFCM). These microspheres provide sustained release of bioactive factors and can be precisely navigated using external magnetic fields to actively penetrate wound barriers, overcoming the limitation of passive diffusion [93].
- Cryogel Delivery Systems: Macroporous chitosan-based cryogels can be used for the controlled and sequential release of factor combinations. For instance, releasing IL-10 and TGF-β early to modulate inflammation, followed by VEGF and FGF to promote angiogenesis and tissue regeneration, has been shown to significantly accelerate wound healing [91].
6. Conclusion
The analysis of the MSC secretome represents a frontier in understanding and harnessing the full therapeutic potential of these cells. For research focused on cryopreservation, rigorous characterization of the secretome is not optional but essential. It serves as a highly sensitive biomarker for cryo-injury, revealing functional deficits that are not apparent from cell viability alone. By employing the standardized production protocols, sophisticated analytical techniques, and engineered delivery systems outlined in this guide, researchers can advance the field towards cryopreservation strategies that truly preserve the functional, secretory integrity of MSCs, ensuring their efficacy in clinical applications.
Within the context of MSC cryopreservation research, optimizing protocols to minimize cryo-injury is paramount for maintaining the therapeutic potential of transplanted cells. A critical step in validating these optimized protocols is demonstrating the in vivo functional efficacy of the post-thaw MSCs in relevant disease models. This guide details the experimental frameworks and quantitative assessments used to evaluate the performance of cryopreserved cellular therapeutics, with a specific focus on models of osteoarthritis. The data presented serve as a benchmark for the functional quality of cells post-preservation, directly reflecting the success of cryopreservation methodologies in mitigating cryo-injury and preserving bioactivity.
Quantitative Efficacy in Osteoarthritis Models
The therapeutic efficacy of interventions, including those potentially involving cellular therapies, is often evaluated in controlled trials using standardized outcome measures. The following table summarizes quantitative findings from key studies on cryotherapy, a common physical intervention, in knee osteoarthritis (KOA), providing a reference point for the magnitude of therapeutic effect in this model system [95] [96].
Table 1: Quantitative Efficacy of Cryotherapy-Based Interventions in Knee Osteoarthritis Models
| Study (Country) | Study Design & Groups | Intervention Protocol | Primary Outcome: Pain | Secondary Outcome: Function | Conclusion on Efficacy |
|---|---|---|---|---|---|
| Mohammedsadiq & Rasool, 2023 (Iraq) [95] [96] | RCT, N=34EG: HBE + Cryotherapy (n=18)CG: HBE only (n=16) | Home-based exercise for 2 months, with EG receiving additional cryotherapy. | WOMAC Pain Score (0-4):Significant improvement in EG. | WOMAC Function Score (0-4):Significant improvement in EG. | Combination of HBE and cryotherapy effectively improves function in KOA patients [95]. |
| Dantas et al., 2019 (Brazil) [95] | RCT, N=60EG: Cryotherapy + Compression (n=30)CG: Sham Cryotherapy (n=30) | Cryotherapy combined with compression and therapeutic exercises for 6 days. | VAS (0-10):No significant reduction in pain in EG. | WOMAC (0-96):Uncertain effects on physical function. | No significant pain reduction; effects on function remain uncertain [95]. |
| Systematic Review & Meta-Analysis (2025) [95] | Meta-analysis of 5 RCTs | Various protocols of cryotherapy, often combined with kinesiotherapy. | Standardized Mean Difference (SMD): -0.57 (95% CI: -0.97 to -0.18; p=0.004). | SMD for functionality (4 studies): -0.28 (95% CI: -0.58 to 0.02; p=0.07). | Cryotherapy is useful as part of comprehensive treatment, especially with kinesiotherapy [95]. |
Detailed Experimental Protocols for In Vivo Evaluation
Protocol for a Randomized Controlled Trial in Knee Osteoarthritis
This protocol exemplifies a robust design for evaluating a non-pharmacological intervention in a KOA model [96].
- Population (P): Adults (40-70 years) diagnosed with KOA (grade ≤3), experiencing knee pain most days of the previous month, with an average pain intensity between 3-7 on a Visual Analog Scale (VAS). Key exclusion criteria include rheumatoid arthritis, lower limb deformities/surgery, uncontrolled diabetes, and contraindications to cryotherapy [96].
- Intervention (I) & Control (C):
- Experimental Group: Undergoes a home-based conventional exercise (HBE) program combined with cryotherapy application for a period of two months [96].
- Control Group 1: Receives the identical HBE program but without cryotherapy [96].
- Control Group 2: Receives regular therapeutic and physiotherapeutic services at the clinic, serving as a standard-care control [96].
- Outcomes (O):
- Time Points: Assessments are conducted at baseline (T0), immediately post-intervention (e.g., 2 months, T1), and at a follow-up visit (e.g., 3 months, T2) to evaluate effect sustainability [95] [96].
Protocol for Assessing Anti-Inflammatory Effects
This protocol is relevant for investigating the mechanistic pathways of cryotherapy or the immunomodulatory effects of cellular therapies like MSCs [97].
- Population: Human subjects, such as athletes or obese individuals, with no major recent illnesses, medications, or unhealthy habits. The focus is on measuring systemic inflammatory markers [97].
- Intervention: Whole-Body Cryotherapy (WBC). Subjects are exposed to extremely low temperatures (typically -110°C to -140°C) for brief durations (1-3 minutes) in multiple sessions over a defined period [97].
- Control: A control group that does not receive the WBC intervention but is otherwise treated identically [97].
- Outcomes:
- Sample Analysis: Blood samples are collected pre- and post-intervention. Serum is isolated, and cytokine levels are quantified using standardized immunoassays (e.g., ELISA) [97].
- Meta-analysis Integration: Data from multiple RCTs can be pooled. For IL-1β, a significant reduction is observed (SMD = -2.08 pg/mL), and for IL-10, a significant increase is observed (SMD = 0.78 pg/mL), indicating a potent anti-inflammatory effect [97].
Signaling Pathways and Experimental Workflows
Anti-inflammatory Signaling Pathway of Cryotherapy
The following diagram illustrates the hypothesized physiological and molecular mechanisms underlying the anti-inflammatory effects observed with cryotherapy, which may mirror pathways leveraged by cellular therapies.
In Vivo Therapeutic Efficacy Workflow
This workflow outlines the sequential phases for evaluating the therapeutic efficacy of an intervention in a disease model, from subject preparation to data synthesis.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for In Vivo Efficacy Studies
| Item / Reagent | Function / Application | Specific Examples / Notes |
|---|---|---|
| WOMAC Index | A standardized, self-administered questionnaire to assess pain, stiffness, and physical function in patients with knee and hip osteoarthritis. | Validated outcome measure; uses subscales for pain (0-20), stiffness (0-8), and physical function (0-68) [95] [96]. |
| Visual Analog Scale (VAS) | A psychometric instrument to measure subjective pain intensity. Patients mark a point on a 10 cm line representing their pain level. | Ranges from "no pain" (0) to "worst pain imaginable" (10) [95]. |
| Enzyme-Linked Immunosorbent Assay (ELISA) Kits | To quantitatively measure concentrations of specific cytokines and inflammatory markers (e.g., IL-1β, IL-10, TNF-α) in serum or plasma samples. | Critical for evaluating mechanistic biomarkers of inflammation and therapeutic response [97]. |
| Whole-Body Cryotherapy (WBC) Chamber | To administer controlled, whole-body cold exposure as an experimental intervention for studying anti-inflammatory and analgesic effects. | Typically operates at -110°C to -140°C with exposure times of 1-3 minutes [97]. |
| Statistical Analysis Software | To perform meta-analysis and data synthesis from multiple randomized controlled trials (RCTs). Calculates effect sizes like Standardized Mean Difference (SMD). | Software like RevMan is standard for Cochrane reviews; used for pooling data and assessing heterogeneity (I² statistic) [95] [97]. |
| PEDro Scale & GRADE Framework | Tools for assessing the methodological quality (risk of bias) of individual studies and the overall certainty of evidence across studies in a systematic review. | PEDro scores RCTs on a 10-point scale. GRADE classifies evidence as high, moderate, low, or very low quality [97]. |
Conclusion
The successful cryopreservation of MSCs hinges on a delicate balance, requiring precise management of the interrelated mechanisms of osmotic, mechanical, and oxidative cryo-injury. While established methods like slow freezing remain robust, the field is advancing towards optimized protocols that incorporate reduced DMSO concentrations, advanced CPAs, and strategic pre-conditioning to better preserve cellular function. Critically, validation must extend beyond simple viability metrics to confirm the retention of therapeutic potency—including immunomodulation, differentiation capacity, and secretome profile—which can be context-dependent for specific clinical applications. Future research must focus on standardizing these optimized protocols for clinical-grade manufacturing, developing novel, less toxic cryoprotectant cocktails, and conducting rigorous comparative studies to definitively map the functional fidelity of cryopreserved MSCs across a broader spectrum of disease models. This will be paramount for realizing the full potential of 'off-the-shelf' MSC therapies in regenerative medicine.
References
- 1. Mesenchymal stem cells in treating human diseases [https://www.nature.com/articles/s41392-025-02313-9]
- 2. Cryopreservation of Human Mesenchymal Stem Cells for ... [https://pubmed.ncbi.nlm.nih.gov/26280501/]
- 3. Effects, methods and limits of the cryopreservation on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11441116/]
- 4. Dimethyl sulfoxide in cryopreserved mesenchymal stromal cell ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06807-6]
- 5. Cryopreservation of Mesenchymal Stem Cells Using ... [https://www.mdpi.com/1422-0067/21/22/8579]
- 6. Identification of a fundamental cryoinjury mechanism in ... [https://www.sciencedirect.com/science/article/pii/S0011224023000913]
- 7. Function of Cryopreserved MSCs with and without IFN-γ pre ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5397371/]
- 8. The mechanism and protective strategies of follicle injury ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12487330/]
- 9. Short-Term Cryopreservation Preserved the Function of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12524150/]
- 10. Effects, methods and limits of the cryopreservation on ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-03954-3]
- 11. Cryopreservation as a Key Element in the Successful ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2020.592242/full]
- 12. Cryopreservation: An Overview of Principles and Cell- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7995302/]
- 13. Osmotic behaviour of human mesenchymal stem cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC5590898/]
- 14. What Is Hydration on a Cellular Level and Why Is It ... [https://www.nutritionnews.abbott/healthy-living/diet-wellness/What-Is-Hydration-on-a-Cellular-Level-and-Why-Is-It-Important/]
- 15. Identification of a fundamental cryoinjury mechanism in MSCs ... [https://pubmed.ncbi.nlm.nih.gov/37827209/]
- 16. Enhanced cryopreservation of MSCs in microfluidic ... [https://www.nature.com/articles/srep35416]
- 17. Principles and Protocols For Post-Cryopreservation Quality ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.907943/full]
- 18. Intracellular Ice Formation: The Enigmatic Role of Cell- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3824294/]
- 19. Mechanisms of intracellular ice formation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1280746/]
- 20. Relation of ice formation to ultrastructural cryoinjury and ... [https://www.sciencedirect.com/science/article/abs/pii/001122407690002X]
- 21. Mechanism of cryoinjury in biological systems [https://www.sciencedirect.com/science/article/pii/0011224072900302]
- 22. The Oxidative Stress of Human Sperm Cryopreservation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12024095/]
- 23. The roles of reactive oxygen species and antioxidants in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6712439/]
- 24. Cryoprotective effects of mesenchymal stem cell and ... [https://www.sciencedirect.com/science/article/abs/pii/S0093691X25002067]
- 25. Melatonin Inhibits Oxidative Stress and Apoptosis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7403229/]
- 26. Cryostorage of Mesenchymal Stem Cells and Biomedical ... [https://www.mdpi.com/2073-4409/11/17/2691]
- 27. Cryostorage of Mesenchymal Stem Cells and Biomedical Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9454587/]
- 28. Advanced cryopreservation engineering strategies: the critical ... [https://cellregeneration.springeropen.com/articles/10.1186/s13619-023-00173-8]
- 29. Mechanisms of cryoinjury in living cells - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/11123179/]
- 30. Ice Inhibition for Cryopreservation: Materials, Strategies, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7967093/]
- 31. Overcoming ice: cutting-edge materials and advanced ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03265-6]
- 32. Cryopreservation based on freezing protocols for the long ... [https://www.sciencedirect.com/science/article/abs/pii/S0142961209002701]
- 33. Cryopreservation of tissues by slow-freezing using an ... [https://www.nature.com/articles/s41598-022-23913-3]
- 34. Technologies for Vitrification Based Cryopreservation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10215456/]
- 35. Genetic Suppression of Cryoprotectant Toxicity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7001869/]
- 36. Vitrification of 3D-MSCs encapsulated in GelMA hydrogel [https://www.sciencedirect.com/science/article/pii/S014181302500265X]
- 37. Cryoprotectant Optimization for Biopharma Stability [https://allanchem.com/cryoprotectant-optimization-for-biopharma-stability/]
- 38. Cryopreservation - Key Survey Findings from the ISCT ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/05/28/cryopreservation-key-survey-findings-from-the-isct]
- 39. Natural Cryoprotective and Cytoprotective Agents in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145333/]
- 40. Chemical approaches to cryopreservation [https://www.nature.com/articles/s41570-022-00407-4]
- 41. Trehalose in cryopreservation. Applications, mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11411628/]
- 42. Cryoprotective Agent - an overview [https://www.sciencedirect.com/topics/nursing-and-health-professions/cryoprotective-agent]
- 43. Cryoprotection synergism between glycerol and dimethyl ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5101303/]
- 44. Review of non-permeating cryoprotectants as supplements ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224020302935]
- 45. Bio-based cryoprotectants, mechanisms, and advanced ... [https://www.sciencedirect.com/science/article/pii/S0924224425003000]
- 46. Cryopreservation of mesenchymal stem/stromal cells using ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324924007734]
- 47. Optimizing mesenchymal stem cell therapy: from isolation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12054297/]
- 48. Low-DMSO Cryopreservation of MSCs with Optibumin 25 ... [https://invitria.com/resources/low-dmso-cryopreservation-of-mscs-with-optibumin-25-recombinant-albumin/]
- 49. Cryopreservation Media - Specialty Cell Culture Media [https://fujifilmbiosciences.fujifilm.com/us/cell-culture-and-processing/specialty-cell-culture-media/cryopreservation-media.html]
- 50. Powering China's Stem Cell Therapy Revolution in 2025 [http://www.cellstore-global.com/news/cellstore-stem-cell-cryopreservation-medium-powering-chinas-stem-cell-therapy-revolution-in-2025.html]
- 51. Cryopreserved Mesenchymal Stromal Cells Maintain ... [https://www.nature.com/articles/srep26463]
- 52. Reconstitution and post-thaw storage of cryopreserved ... [https://www.sciencedirect.com/science/article/pii/S2352320423000275]
- 53. Dimethyl Sulfoxide-Free Cryopreservation of Human Umbilical Cord... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8306716/]
- 54. Priming approaches to improve the efficacy of mesenchymal ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-019-1224-y]
- 55. Quantitative assessment of the impact of cryopreservation on ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-020-02054-2]
- 56. Screening for cryoprotective agent toxicity and ... [https://pubmed.ncbi.nlm.nih.gov/40992110/]
- 57. Innovations in Platelet Cryopreservation: Evaluation of DMSO ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563202/]
- 58. Therapeutic Potential and Mechanisms of Mesenchymal Stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12156323/]
- 59. Cryopreservation in Cell and Gene Therapy [https://cgt.global/cryopreservation-in-cell-and-gene-therapy-ensuring-high-viability-cells-for-research-manufacturing/]
- 60. Interrupted cooling protocols in cryopreservation: A review ... [https://pubmed.ncbi.nlm.nih.gov/41192344/]
- 61. Hydrogel microcapsulation technology reduces the DMSO ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12639263/]
- 62. Strategies to enhance stability of cryopreservation ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002496]
- 63. Short-Term Cryopreservation Preserved the Function of ... [https://www.mdpi.com/2073-4409/14/19/1569]
- 64. A Closer Look at the Potential Mechanisms of Action ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11858741/]
- 65. Quantitative phosphoproteomics explain cryopreservation ... [https://www.sciencedirect.com/science/article/abs/pii/S187439192400085X]
- 66. The Utilization of Freezing Steps in Mesenchymal Stromal ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.01627/full]
- 67. IFN-γ and TNF-α Pre-licensing Protects Mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5910660/]
- 68. Cryopreservation does not change the performance and ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-022-03198-z]
- 69. A non-traditional approach to cryopreservation by ultra-rapid ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0220055]
- 70. Impact of Cryopreservation and Freeze-Thawing on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9045030/]
- 71. Key quality parameter comparison of mesenchymal stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11324487/]
- 72. Thawed Mesenchymal Stem Cell Product Shows ... [https://www.nature.com/articles/s41598-019-54462-x]
- 73. Optimisation of cryopreservation conditions, including storage ... [https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/s12860-024-00516-6]
- 74. Comparison of freshly cultured versus cryopreserved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9286731/]
- 75. Quantitative assessment of the impact of cryopreservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7734731/]
- 76. Key quality parameter comparison of mesenchymal stem ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1412811/full]
- 77. Cryopreserved mesenchymal stem cells regain functional ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-019-2038-5]
- 78. Exploring the Potential Effects of Cryopreservation on the ... [https://www.mdpi.com/1422-0067/25/18/9908]
- 79. Cryopreservation of Human Mesenchymal Stem Cells in an ... [https://www.nature.com/articles/s41598-017-16134-6]
- 80. Adipose-derived mesenchymal stem cells (MSCs) are a ... [https://www.sciencedirect.com/science/article/pii/S2452199X23003997]
- 81. Donor age effects on in vitro chondrogenic and osteogenic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9632053/]
- 82. Adipogenic Potential of Adipose Stem Cell Subpopulations [https://pmc.ncbi.nlm.nih.gov/articles/PMC4167367/]
- 83. Osteogenic commitment of Wharton's jelly mesenchymal ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-019-1450-3]
- 84. Mesenchymal Stem Cells from Fat: From Differentiation ... [https://www.intechopen.com/chapters/1190125]
- 85. Assessing Adipogenic Potential of Mesenchymal Stem Cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC3574742/]
- 86. Adipogenesis, Osteogenesis, and Chondrogenesis of Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7362937/]
- 87. Mesenchymal Stem Cell Immunomodulation: Mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7751844/]
- 88. The Role of Indoleamine 2, 3-Dioxygenase in Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4586474/]
- 89. Immune regulation through tryptophan metabolism [https://www.nature.com/articles/s12276-023-01028-7]
- 90. Mesenchymal stem cell secretome for regenerative medicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC11976416/]
- 91. Sequential Delivery of Cryogel Released Growth Factors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7212449/]
- 92. Method standardization of secretome production, collection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12423684/]
- 93. Stem cell secretome armed magneto-actuated micromotors ... [https://www.nature.com/articles/s41467-025-61914-8]
- 94. The Pattern of Cytokines, Chemokines, and Growth Factors ... [https://www.mdpi.com/2077-0383/13/23/7064]
- 95. Cryotherapy in Knee Osteoarthritis: A Systematic Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12168428/]
- 96. Effectiveness of home-based conventional exercise and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10158910/]
- 97. Whole-body cryotherapy can reduce the inflammatory ... [https://www.nature.com/articles/s41598-025-90396-3]