Oncogenic Potential in Stem Cells: Assessment Strategies and Risk Mitigation for Research and Therapy
This article provides a comprehensive analysis of oncogenic risk assessment across diverse stem cell types, including pluripotent stem cells (PSCs), induced pluripotent stem cells (iPSCs), and adult stem cells.
Oncogenic Potential in Stem Cells: Assessment Strategies and Risk Mitigation for Research and Therapy
Abstract
This article provides a comprehensive analysis of oncogenic risk assessment across diverse stem cell types, including pluripotent stem cells (PSCs), induced pluripotent stem cells (iPSCs), and adult stem cells. It explores the fundamental biological mechanisms driving tumorigenicity, details current and emerging methodologies for risk evaluation, addresses key challenges in safety profiling, and presents comparative frameworks for validating assessment strategies. Tailored for researchers, scientists, and drug development professionals, this review synthesizes the latest advances in the field to inform safer therapeutic development and more robust preclinical screening protocols.
Understanding the Roots of Risk: Stem Cell Biology and Tumorigenic Mechanisms
Stem cells represent a cornerstone of regenerative medicine due to their unique capacities for self-renewal and differentiation. However, these very properties are also hallmarks of cancer, creating a critical challenge for therapeutic development. The oncogenic potential—the ability to initiate tumor formation—varies significantly across different stem cell types and is influenced by distinct molecular mechanisms. Understanding these differences is paramount for advancing safe and effective stem cell-based therapies. This guide provides a systematic comparison of the oncogenic risk profiles of three major stem cell categories: pluripotent stem cells (PSCs), including both embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), and adult stem cells (ASCs). For researchers and drug development professionals, this comparison is essential for making informed decisions about model system selection, risk mitigation strategies, and clinical translation pathways.
Comparative Oncogenic Risk Profiles of Major Stem Cell Types
Table 1: Oncogenic Risk Profile by Stem Cell Type
| Stem Cell Type | Key Oncogenic Risks | Primary Tumor Types | Contributing Factors |
|---|---|---|---|
| Induced Pluripotent Stem Cells (iPSCs) | - Teratoma formation from undifferentiated cells- Malignant transformation from partially reprogrammed cells- Reactivation of reprogramming factors (e.g., c-Myc) [1] | Teratomas, somatic tumors | Genomic integration of vectors, oncogenic transgenes, incomplete reprogramming, epigenetic aberrations [2] [1] |
| Embryonic Stem Cells (ESCs) | - Teratoma formation from residual undifferentiated cells [1] | Teratomas | Spontaneous differentiation, genomic instability during long-term culture |
| Adult Stem Cells (ASCs) / Cancer Stem Cells (CSCs) | - Initiation of primary tumors- Therapy resistance and cancer recurrence [3] | Carcinomas, leukemias, solid tumors | Accumulated mutations in long-lived cells, aberrant niche signaling, epigenetic dysregulation [4] [3] |
Table 2: Key Molecular Drivers and Markers of Oncogenesis
| Stem Cell Type | Core Pluripotency/Ongenic Drivers | Characteristic Markers | Regulatory Pathways |
|---|---|---|---|
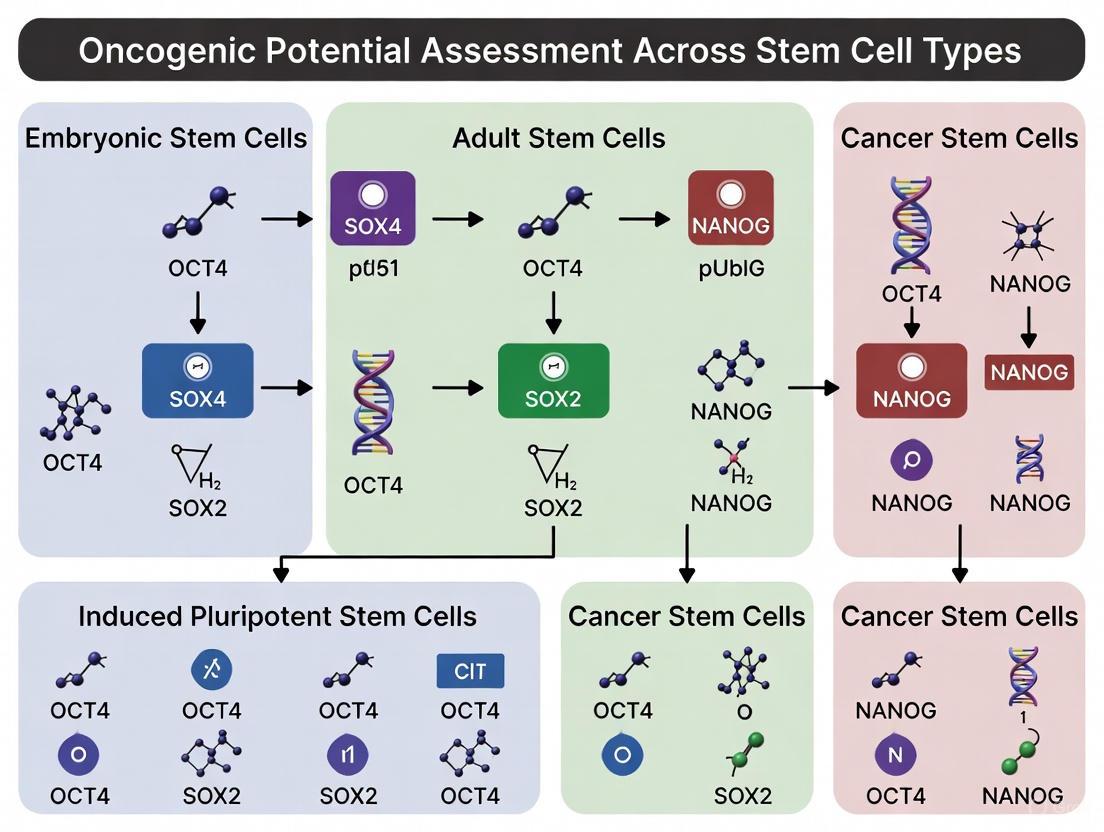
| PSCs (iPSCs/ESCs) | Oct4, Sox2, Nanog, c-Myc, Klf4 [1] | Alkaline Phosphatase, SSEA-4, TRA-1-60, TRA-1-81 | Wnt/β-Catenin, Myc-centered network [1] |
| Adult Stem Cells (ASCs) | Varies by tissue type | Varies by tissue (e.g., Lgr5 for intestine, CD34+CD38- for hematopoiesis) | Notch, Hedgehog, Wnt [4] |
| Cancer Stem Cells (CSCs) | Often Oct4, Sox2, Nanog (re-activated) [3] | CD44+CD24- (breast), CD133+ (brain, colon), CD34+CD38- (AML) [3] | Wnt/β-Catenin, Hedgehog, Notch, JAK/STAT [3] |
Experimental Models and Assays for Assessing Oncogenic Potential
In Vitro Transformation Assays
The oncogenic foci (OF) formation assay is a classical method to study transformation. Strikingly, the methodological parallels between OF production and iPSC generation are profound; both involve transducing fibroblasts with specific sets of genes and result in colonies with distinct morphologies [5]. Direct comparison of the transcriptomes of iPSCs and OF derived from common parental mouse embryonic fibroblasts (MEFs) revealed substantial overlap, including shared downregulation of differentiation-associated genes and upregulation of monosaccharide metabolism pathways [5]. However, a critical distinction lies in the specific activation of a 17-gene pluripotency cluster in iPSCs that is absent in OF, underscoring that while the processes are related, the cell types are distinct [5].
In Vivo Tumorigenicity Testing
The gold-standard assay for validating the pluripotency of PSCs—teratoma formation—is simultaneously a demonstration of their oncogenic potential. Upon injection into immunodeficient mice, undifferentiated PSCs form teratomas, benign tumors containing tissues from all three germ layers [1]. The risk extends beyond teratomas; studies have documented the formation of neural overgrowths and ocular tumors from transplanted human ESC-derived dopaminergic neurons and retinal progenitors in animal models [1]. To assess the tumor-initiating capacity of CSCs, researchers employ xenotransplantation models using immunodeficient mice (e.g., NOD/SCID strains). The frequency of CSCs is quantified by limiting dilution assays, which measure the cell dose required to form a tumor upon serial transplantation [3].
Molecular Mechanisms Governing Oncogenic Transformation
Shared Transcriptional Networks in Pluripotency and Cancer
A foundational link between PSCs and cancer is the shared activity of core transcriptional networks. Myc and the core pluripotency circuit (Nanog, Oct4, and Sox2) are fundamental to both pluripotency maintenance and oncogenesis, promoting self-renewal, proliferation, and resistance to differentiation [1]. Bioinformatics analyses reveal that more aggressive cancers often exhibit high expression of these core pluripotency and Myc-centered networks [1]. The ectopic expression of these factors during iPSC reprogramming, particularly the integration and potential reactivation of the potent oncogene c-Myc, presents a significant and well-documented tumorigenic risk [1].
Diagram Title: Shared Transcriptional Networks in Pluripotency and Cancer
Signaling Pathways in Cancer Stem Cells and Niches
In adult tissues, CSCs and normal ASCs reside in specialized niches that regulate their behavior through key developmental signaling pathways. Disruption of this crosstalk is a common mechanism in oncogenesis. The Wnt/β-Catenin, Hedgehog, Notch, NF-κB, JAK/STAT, and TGF-β pathways are frequently dysregulated in CSCs, contributing to their maintenance, self-renewal, and therapeutic resistance [3]. The origin of CSCs is context-dependent, with evidence suggesting they can arise from normal ASCs that accumulate mutations or from more differentiated progenitor cells that re-acquire self-renewal capacity through epigenetic or mutational events [4]. The long lifespan and inherent self-renewal capacity of ASCs make them particularly susceptible to accumulating the genetic "hits" required for transformation [4].
Diagram Title: Key Pathways and CSCs
Research Reagent Solutions for Oncogenicity Studies
Table 3: Essential Research Toolkit for Oncogenic Potential Assessment
| Reagent / Tool Category | Specific Examples | Research Application |
|---|---|---|
| Reprogramming Vectors | Excisable lentivirus (doxycycline-inducible), PiggyBac transposon, Sendai virus, mRNA [1] | Generating footprint-free iPSCs; minimizing genomic integration risks. |
| Cell Sorting Markers | Antibodies against CD34, CD38, CD44, CD24, CD133, ALDH activity assays [3] | Isulating and purifying putative CSCs and normal ASC populations for functional study. |
| Pathway Modulators | Small molecule inhibitors of Wnt (e.g., IWP-2), Hedgehog (e.g., cyclopamine), Notch (e.g., DAPT) [3] | Probing the functional contribution of specific signaling pathways to stem cell maintenance and transformation. |
| In Vivo Models | Immunodeficient mice (e.g., NOD/SCID, NSG) | Conducting teratoma and tumorigenicity assays via xenotransplantation. |
The landscape of oncogenic potential across stem cell types is complex and multifaceted. PSCs, particularly iPSCs, carry a significant risk of teratoma formation and transformation driven by their core pluripotency networks and the technical challenges of reprogramming. In contrast, the primary risk associated with ASCs is their susceptibility to serve as the cell of origin for cancers over time due to their longevity. The emergence of CSCs across numerous malignancies presents a major therapeutic challenge in oncology, driving disease recurrence and resistance.
Future progress hinges on continued refinement of safety measures. For iPSCs, this includes the adoption of non-integrating reprogramming methods, advanced purification techniques to remove undifferentiated cells (e.g., fluorescence-activated cell sorting using pluripotency surface markers), and the development of "suicide genes" as a safety switch in cell therapies [1]. In the realm of CSCs, the focus is on identifying novel, specific surface markers and understanding the dynamic plasticity that allows non-CSCs to re-acquire stem-like properties, paving the way for more effective combinatorial therapies that can prevent relapse [3]. A deep understanding of these distinct oncogenic risk profiles is indispensable for guiding the safe clinical application of stem cell technologies and for developing novel, curative strategies against cancer.
The therapeutic promise of stem cells in regenerative medicine is underpinned by three core mechanistic properties: self-renewal, differentiation capacity, and genetic stability. These interdependent functions determine both the therapeutic potential and the inherent oncogenic risks of any stem cell population. Self-renewal refers to the ability of a stem cell to divide and produce identical copies of itself, thereby maintaining the stem cell pool throughout life. Differentiation capacity (potency) defines the range of specialized cell types a stem cell can generate, ranging from pluripotent (all embryonic lineages) to multipotent (limited to tissue-specific lineages). Genetic stability ensures the faithful transmission of genomic information during cell division, preventing the accumulation of mutations that could lead to malignant transformation [6] [7].
The precise regulation of self-renewal pathways is critical, as their dysregulation is a hallmark of cancer. Cancer stem cells (CSCs), a subpopulation within tumors, co-opt these same pathways to drive tumor initiation, maintenance, metastasis, and therapy resistance. Consequently, a comparative assessment of these core mechanisms across stem cell types is not merely an academic exercise but a fundamental prerequisite for evaluating their therapeutic safety profile and oncogenic potential [8] [9].
Self-Renewal Pathways: Mechanisms and Dysregulation
Self-renewal in stem cells is governed by a set of evolutionarily conserved signaling pathways. In normal stem cells, these pathways are tightly regulated; however, in CSCs, they are often deregulated, leading to unchecked proliferation.
Key Signaling Pathways in Self-Renewal
The principal pathways governing stem cell self-renewal include Hedgehog (Hh), Wnt, Notch, and the B-cell-specific moloney murine leukemia virus integration site 1 (BMI1) pathway [9].
- Hedgehog (Hh) Pathway: In the absence of the Hh ligand, the Patched (Ptc) receptor inhibits Smoothened (Smo), preventing activation of the Gli transcription factors. Upon Hh binding, this inhibition is relieved, leading to Gli-mediated transcription of target genes (e.g., cyclins) that promote self-renewal. This pathway is crucial in CSCs of cancers like basal cell carcinoma, leukemia, and pancreatic cancer [9].
- Wnt Pathway: In a canonical Wnt-off state, a destruction complex targets β-catenin for degradation. Wnt signaling stabilizes β-catenin, allowing its translocation to the nucleus to activate genes like c-MYC and CYCLIN D1, which drive proliferation and self-renewal. Aberrant Wnt signaling is implicated in various CSCs [9].
- Notch Pathway: Notch signaling is initiated by ligand-receptor (e.g., Delta, Jagged) interaction between adjacent cells. This triggers proteolytic cleavage of the Notch intracellular domain (NICD), which translocates to the nucleus and activates target genes such as Hairy and enhancer of split (HES) family members. Dysregulated Notch signaling contributes to T-cell acute lymphoblastic leukemia (T-ALL) and breast CSCs [9].
- Transcriptional Networks in Squamous Cell Carcinoma (SCC): A specific bi-stable network involving PITX1, SOX2, and TP63 promotes self-renewal and suppresses differentiation in SCC. PITX1 and SOX2, which are epigenetically repressed in normal skin, become expressed in SCC tumor-propagating cells (TPCs). They cooperatively bind enhancers to activate pro-proliferation genes and repress the differentiation factor KLF4, maintaining the TPC state [10].
The following diagram illustrates the core logic of these self-renewal pathways and their frequent dysregulation in CSCs.
Comparative Self-Renewal Mechanisms Across Stem Cell Types
Different stem cell types exhibit distinct self-renewal behaviors and regulatory mechanisms, which directly influence their oncogenic potential. The following table compares these aspects across major stem cell categories.
Table 1: Comparative Analysis of Self-Renewal and Oncogenic Risk Across Stem Cell Types
| Stem Cell Type | Self-Renewal Capacity | Key Regulatory Pathways & Factors | Oncogenic Potential & Associated Risks |
|---|---|---|---|
| Embryonic Stem Cells (ESCs) [6] [11] | High, unlimited self-renewal in vitro; pluripotent. | Oct4, Nanog, Sox2; dependent on specific growth factors (FGF, TGF-β). | High teratoma risk in vivo due to pluripotency; requires precise pre-differentiation [11] [7]. |
| Induced Pluripotent Stem Cells (iPSCs) [6] | High, similar to ESCs; pluripotent. | Reprogramming factors (Oct4, Sox2, Klf4, c-Myc); same pathways as ESCs. | Risk from reprogramming methods (e.g., viral integration); potential for epigenetic abnormalities [6]. |
| Adult/Mesenchymal Stem Cells (MSCs) [6] [12] | Limited in vitro expansion; multipotent. | Microenvironment (niche) dependent; express CD73, CD90, CD105. | Lower tumorigenicity; primary risk is genomic instability during long-term culture [7] [12]. |
| Hematopoietic Stem Cells (HSCs) [6] [13] | Lifelong, balanced self-renewal and differentiation. | Tightly regulated by bone marrow niche; transcription factors (EVI1). | Imbalance linked to hematological malignancies; aging increases self-renewal bias [13]. |
| Cancer Stem Cells (CSCs) [8] [9] | Deregulated, excessive self-renewal. | Dysregulated Hh, Wnt, Notch, BMI1; SCC-specific: PITX1-SOX2-TP63 network [10]. | Directly drives tumorigenesis, metastasis, and therapy resistance. |
Assessing Differentiation Capacity and Genetic Stability
The differentiation potential of a stem cell is a double-edged sword, offering regenerative capability while posing a risk of uncontrolled growth if differentiation fails.
Differentiation Capacity and Its Therapeutic Implications
- Pluripotency and Lineage Specification: ESCs and iPSCs can differentiate into derivatives of all three embryonic germ layers (ectoderm, mesoderm, endoderm), making them powerful for disease modeling and generating diverse cell types for therapy. However, this broad potential necessitates rigorous in vitro differentiation protocols to ensure no undifferentiated cells remain in therapeutic products, as these can cause teratomas [6] [11].
- Multipotency and Tissue Repair: Adult stem cells, including MSCs and HSCs, are multipotent, with differentiation restricted to lineages of their tissue of origin. MSCs can differentiate into osteoblasts, chondrocytes, and adipocytes, and their therapeutic effect is largely mediated via paracrine signaling and immunomodulation, reducing the risk of aberrant differentiation [6] [12].
- Blocked Differentiation in Cancer: A hallmark of CSCs is their ability to self-renew excessively while failing to undergo terminal differentiation. The PITX1-SOX2-TP63 network in SCCs, for example, actively suppresses the differentiation factor KLF4, locking cells in a proliferative, stem-like state [10].
Genetic Stability Assessment and Protocols
Maintaining genetic integrity is paramount for safe clinical application. The following experimental protocols are central to biosafety assessment.
- Karyotyping and Genetic Analysis: This is a standard method for detecting gross chromosomal abnormalities (e.g., aneuploidy, translocations) that may arise during long-term culture of stem cells like ESCs and iPSCs. It involves arresting cells in metaphase, staining the chromosomes, and analyzing their number and structure [7].
- In Vivo Tumorigenicity Assay: This is the definitive test for assessing the potential of a stem cell product to form tumors. The protocol involves immunocompromised mice (e.g., NOD/SCID) [7] [8].
- Cell Preparation: The stem cell product is prepared at the intended clinical dose and higher doses.
- Administration: Cells are implanted into mice via a clinically relevant route (e.g., subcutaneous, intramuscular).
- Observation: Animals are monitored for an extended period (e.g., 6 months) for signs of tumor formation.
- Necropsy and Histopathology: At the endpoint, the implantation site and major organs are examined grossly and microscopically for any neoplastic growths or teratomas.
- Oncogenicity/Teratogenicity Testing: This assesses the potential of cells, particularly pluripotent ones, to cause tumors or disruptive embryonic growth. It uses a combination of in vitro methods and in vivo models in immunocompromised animals, evaluating the formation of complex, disorganized tissues indicative of teratoma formation [7].
The workflow for a comprehensive biosafety assessment integrates multiple of these experimental approaches, as shown below.
The Scientist's Toolkit: Key Reagents and Research Solutions
Advancing research in stem cell self-renewal and oncogenic potential requires a suite of specialized reagents and tools. The following table details essential solutions for key experimental workflows.
Table 2: Key Research Reagent Solutions for Stem Cell and CSC Mechanism Studies
| Research Reagent / Tool | Primary Function in Research | Key Applications |
|---|---|---|
| Small Molecule Pathway Inhibitors [9] | Chemically inhibit key self-renewal pathway components (e.g., Smo for Hh, γ-secretase for Notch). | Functional validation of pathway necessity in CSCs; target identification for therapeutic development. |
| CRISPR-Cas9 Systems [6] | Enable precise genome editing for gene knockout, knock-in, or mutation. | Functional screens to identify self-renewal genes; introduce or correct oncogenic mutations; generate disease models. |
| Flow Cytometry Antibodies (CD44, CD133, CD73, CD90, CD105) [8] [12] | Identify and isolate specific stem cell and CSC populations based on surface marker expression. | Phenotypic characterization; purification of homogeneous cell populations for functional assays. |
| scRNA-Seq Kits [6] [8] | Profile gene expression at single-cell resolution. | Deconvolute intra-tumor heterogeneity; identify rare CSC subpopulations; trace lineage commitment. |
| 3D Organoid Culture Systems [8] | Provide a more physiologically relevant, three-dimensional environment for cell growth. | Model tumor-stroma interactions; study CSC dynamics and drug response ex vivo. |
The path from bench to bedside for stem cell therapies is fraught with the challenge of harnessing potent self-renewal and differentiation capacities while mitigating oncogenic risks. A direct comparison reveals that pluripotent cells (ESCs, iPSCs) offer unparalleled therapeutic potential but carry the highest inherent risk of teratoma formation, demanding stringent pre-differentiation and genetic stability checks. In contrast, adult stem cells like MSCs present a lower tumorigenic risk but have limited expansion and differentiation potential. The most critical insight is that the very pathways essential for normal stem cell function—Hedgehog, Wnt, and Notch—are the ones most frequently subverted by CSCs to drive malignancy [6] [9].
Therefore, a rigorous, multi-parametric biosafety assessment is non-negotiable. This includes comprehensive product quality control (sterility, identity, potency), validated tumorigenicity assays in sensitive models, and long-term genetic stability monitoring [7]. Emerging tools like AI-driven multi-omics analysis [8] and proteomic-based stemness indices [14] promise more refined risk stratification. The future of safe and effective stem cell-based therapies lies in an integrated approach that continuously evaluates the delicate balance between regenerative potential and oncogenic danger across all stages of product development.
Cancer Stem Cells (CSCs) represent a functionally distinct subpopulation within tumors that possess the capacity to drive tumor initiation, progression, metastasis, and therapeutic resistance. According to the CSC hypothesis, only a small subset of cancer cells has the ability to recapitulate the formation of a growing tumor [15]. These cells share critical properties with normal stem cells, most notably long-term self-renewal capacity and the ability to differentiate into heterogeneous cancer cell lineages that comprise the tumor bulk [15] [8]. The concept that stemness plays a dual role in both initiating and maintaining tumors has fundamentally transformed our understanding of oncogenesis and presents crucial implications for therapeutic development.
The CSC model contrasts with earlier stochastic models of cancer development, which proposed that most cancer cells possess similar tumorigenic potential [15]. Instead, accumulating evidence supports a hierarchical organization in many tumors, with CSCs sitting at the apex and driving the production of more differentiated daughter cells with limited proliferative potential [15] [8] [16]. This paradigm shift necessitates a reevaluation of oncogenic potential assessment across stem cell types and demands therapeutic strategies that specifically target the CSC population to achieve durable remission and prevent recurrence.
Biological Foundations of Cancer Stem Cells
Historical Context and Theoretical Evolution
The conceptual foundations of CSCs extend back to the 19th century with Rudolf Virchow's dictum "omnis cellula e cellula" (every cell from a cell) and Julius Cohnheim's "embryonal rest hypothesis," which proposed that tumors arise from residual embryonic cells persisting in adult tissues [8]. Modern CSC theory gained substantial experimental support in 1994-1997 with John Edgar Dick's groundbreaking work identifying SCID-leukemia-initiating cells (SL-ICs) in acute myeloid leukemia (AML) characterized by a CD34⁺CD38⁻ phenotype [8]. This established that only a specific cellular subpopulation possessed leukemia-initiating potential, providing the first rigorous experimental evidence for the CSC model in human cancer [8].
Subsequent research has demonstrated that CSCs exist across diverse cancer types, including solid tumors such as glioblastoma, breast cancer, lung cancer, prostate cancer, colon cancer, head and neck squamous cell carcinoma, pancreatic cancer, and melanoma [8]. The CSC hypothesis serves as a supplement rather than a replacement for conventional oncogenic theory, focusing attention on the cell type targeted by molecular events rather than questioning the events themselves [15].
Functional Properties and Defining Characteristics
CSCs are functionally defined by several hallmark characteristics that distinguish them from the bulk tumor population:
Self-Renewal Capacity: CSCs undergo extended self-renewal through mitotic division, maintaining the stem cell pool through either asymmetric or symmetric division [15]. This capacity is thought to be a determining factor in tumor maintenance and regrowth [15].
Differentiation Potential: CSCs can differentiate into the heterogeneous lineages of cancer cells that constitute the tumor, contributing to intratumoral heterogeneity [15] [8].
Therapy Resistance: CSCs demonstrate enhanced resistance to conventional therapies including chemotherapy and radiation, attributed to mechanisms such as enhanced DNA repair systems, drug efflux pumps, and quiescence [8] [17].
Metabolic Plasticity: CSCs can switch between glycolysis, oxidative phosphorylation, and alternative fuel sources such as glutamine and fatty acids, enabling survival under diverse environmental conditions [8].
Interaction with the Tumor Microenvironment: CSCs constantly interact with stromal cells, immune components, and vascular endothelial cells, facilitating metabolic symbiosis that further promotes CSC survival and drug resistance [8] [18].
Table 1: Core Functional Properties of Cancer Stem Cells
| Property | Functional Significance | Therapeutic Implications |
|---|---|---|
| Self-Renewal | Sustains long-term tumor growth and regeneration | Targeting self-renewal pathways may deplete CSC reservoir |
| Differentiation Capacity | Generates cellular heterogeneity within tumors | Differentiation therapy may reduce tumor aggressiveness |
| Therapy Resistance | Leads to treatment failure and relapse | Requires specific CSC-targeting approaches |
| Metabolic Plasticity | Adapts to nutrient deprivation and stress | Dual metabolic inhibition may overcome adaptation |
| Microenvironment Interaction | Creates protective niches | Targeting niche components may sensitize CSCs |
The Dual Role of Stemness: Initiation Versus Maintenance
Stemness in Tumor Initiation: Cellular Origins
The origin of CSCs remains a subject of ongoing investigation, with evidence supporting multiple potential cellular sources. Current research indicates that CSCs may originate from either transformed tissue-resident stem cells or de-differentiated somatic cells that reacquire stem-like properties [16]. The specific cell of origin significantly influences tumor characteristics, prognosis, and aggressiveness [16].
In hematopoietic malignancies, the cellular origin is relatively well-established. Acute myeloid leukemia (AML) typically originates from hematopoietic stem or progenitor cells [16], while chronic myeloid leukemia (CML) is characterized by the Bcr-Abl oncogene resulting from chromosomal translocation between chromosomes 9 and 22 [16]. For solid tumors, the picture is more complex. Gastric cancers may arise from slow-cycling Mist1-expressing cells in the gastric corpus or Lgr5-expressing cells in the gastric antrum [16]. Breast tumors originating from luminal progenitors are generally associated with better prognosis, while those originating from basal-like progenitors display more aggressive phenotypes [16].
The transformation process involves both genetic and epigenetic alterations. Normal stem cells may require fewer genomic changes for transformation since they already possess inherent self-renewal capacity [16]. Differentiated cells, in contrast, must undergo de-differentiation and acquire self-renewal capabilities, potentially through the reactivation of embryonic programs [19]. This process shares remarkable similarities with induced pluripotency, utilizing overlapping molecular signaling and epigenetic pathways [19].
Stemness in Tumor Maintenance: Mechanisms and Pathways
Once tumors are established, stemness properties shift to maintenance functions, promoting continuous growth, fostering heterogeneity, and enabling adaptation to therapeutic pressures. Several evolutionarily conserved signaling pathways play critical roles in maintaining CSC self-renewal and survival:
- Wnt/β-catenin Signaling: Regulates self-renewal in various CSC populations and contributes to therapeutic resistance [15] [19].
- Notch Signaling: Plays essential roles in cell fate decisions and maintains CSC populations across multiple cancer types [15].
- Hedgehog (Hh) Signaling: Contributes to CSC maintenance and tissue patterning [15].
- NF-κB Pathway: Promotes inflammatory responses and supports CSC survival [15].
- BMI1 Polycomb Protein: Regulates self-renewal through epigenetic mechanisms [15].
These pathways operate within a complex regulatory network influenced by both intrinsic genetic programs and extrinsic cues from the tumor microenvironment [8] [18]. The dynamic interaction between CSCs and their niches creates specialized microenvironments that maintain stemness properties through cytokine networks, metabolic symbiosis, and physical interactions [18].
Diagram 1: Signaling Pathways Regulating CSC Stemness Properties. Multiple evolutionarily conserved pathways converge to maintain core CSC functionalities that support both tumor initiation and maintenance.
Experimental Assessment of CSCs: Methodologies and Biomarkers
CSC Identification and Isolation Techniques
Experimental assessment of CSCs relies on a combination of surface marker expression, functional assays, and transplantation models. No single marker is entirely specific for CSCs, requiring multiparameter approaches for reliable identification [15] [19]. The gold standard for validating CSC properties remains the demonstration of tumor-initiating capacity in immunodeficient mouse models, where purified cell populations are transplanted to assess their ability to recapitulate tumor heterogeneity [15].
Table 2: Key Experimental Methods for CSC Identification and Characterization
| Method Category | Specific Techniques | Experimental Readouts | Key Considerations |
|---|---|---|---|
| Surface Marker-Based Isolation | FACS (Fluorescence-Activated Cell Sorting), MACS (Magnetic-Activated Cell Sorting) | Purity of CSC populations, Marker expression profiles | Marker heterogeneity across cancer types, Dynamic expression changes |
| Functional Assays | Sphere Formation, Limiting Dilution Transplantation, Side Population Analysis | Self-renewal capacity, Tumor-initiating frequency, Drug efflux capability | Microenvironment influence, Assay stringency, Technical variability |
| Omics Profiling | Single-Cell RNA Sequencing, Proteomics, Phosphoproteomics, Glycoproteomics | Molecular signatures, Pathway activation, Heterogeneity mapping | Computational complexity, Data integration challenges |
| In Vivo Tracking | Lentiviral barcoding, Bioluminescent imaging, Lineage tracing | CSC dynamics, Clonal evolution, Metastatic patterns | Model organism limitations, Microenvironment differences |
CSC Biomarkers Across Cancer Types
Biomarkers for CSCs include cell surface antigens, intracellular transcription factors, and functional characteristics that vary across different cancer types. These biomarkers facilitate both experimental assessment and therapeutic targeting of CSCs.
Table 3: Established CSC Biomarkers Across Different Cancer Types
| Cancer Type | Key Biomarkers | Associated Functional Properties | Clinical Correlations |
|---|---|---|---|
| Acute Myeloid Leukemia (AML) | CD34⁺CD38⁻ [15] [8] | Chemoresistance, Dormancy | Poor prognosis, Relapse risk |
| Breast Cancer | CD44⁺CD24⁻/low, ALDH1⁺ [8] [19] | Metastasis, Therapy resistance | Shorter recurrence-free survival |
| Glioblastoma | CD133⁺, Nestin⁺, SOX2⁺ [15] [8] | Radioresistance, Invasion | Tumor aggressiveness |
| Colon Cancer | CD133⁺, CD44⁺, LGR5⁺ [8] [19] | Metastasis, Regeneration capacity | Recurrence risk |
| Pancreatic Cancer | CD133⁺, CD44⁺, CXCR4⁺ [8] | Stromal interaction, Drug resistance | Therapeutic resistance |
| Lung Cancer | CD133⁺, ALDH⁺ [8] | Plasticity, Adaptability | Advanced disease stage |
It is important to note that CSC biomarkers demonstrate significant plasticity, altering their expression in response to therapy, microenvironmental cues, and metabolic stress [19]. This dynamic regulation complicates both experimental assessment and therapeutic targeting, necessitating multiparameter approaches that account for CSC heterogeneity and adaptability.
Experimental Protocols for CSC Assessment
Tumor Sphere Formation Assay
The tumor sphere formation assay represents a fundamental functional test for CSC self-renewal and survival under non-adherent conditions. This protocol assesses the capacity of single cells to form clonal non-adherent spheres in serum-free conditions with defined growth factors [8].
Materials and Reagents:
- Ultra-low attachment plates
- Serum-free DMEM/F12 medium
- B27 supplement (50x)
- Recombinant human EGF (20 ng/mL)
- Recombinant human bFGF (10 ng/mL)
- Penicillin-Streptomycin solution
- Accutase enzyme solution for dissociation
Procedure:
- Dissociate tumor samples or cultured cells to single-cell suspension using Accutase
- Filter cells through 40μm strainer to remove aggregates
- Resuspend cells in serum-free sphere medium at 1,000-10,000 cells/mL
- Plate 100-200 μL per well in 96-well ultra-low attachment plates
- Culture for 7-14 days at 37°C with 5% CO₂
- Supplement with fresh growth factors every 3-4 days
- Quantify sphere number and diameter using inverted microscopy
- Passage spheres for serial sphere formation assays to assess self-renewal capacity
Interpretation and Analysis: Sphere-forming efficiency (SFE) is calculated as (number of spheres formed / number of cells seeded) × 100. Primary spheres can be dissociated and replated to assess self-renewal capacity through secondary and tertiary sphere formation. This assay provides a functional readout of CSC frequency and self-renewal potential under defined conditions.
Limiting Dilution Transplantation Assay
The limiting dilution transplantation assay represents the gold standard for quantifying tumor-initiating cell frequency through in vivo transplantation [15] [8]. This method provides the most rigorous assessment of CSC functional properties.
Materials and Reagents:
- Immunocompromised mice (NOD/SCID, NSG)
- Matrigel basement membrane matrix
- Cell sorting equipment (FACS) with appropriate antibodies
- Anesthetic reagents (isoflurane, ketamine/xylazine)
- Sterile surgical instruments for orthotopic transplantation
Procedure:
- Prepare single-cell suspensions from tumor specimens or cell lines
- Sort cells into defined subpopulations based on marker expression
- Serially dilute cells (e.g., 10⁵, 10⁴, 10³, 10², 10 cells) in PBS:Matrigel (1:1)
- Transplant cells orthotopically or subcutaneously into immunocompromised mice
- Monitor tumor formation weekly by palpation or imaging
- Continue observation for 12-24 weeks depending on cancer type
- Sacrifice animals at endpoint or when tumors reach 1.5cm diameter
- Analyze tumor histology to confirm recapitulation of original heterogeneity
Data Analysis: Tumor-initiating cell frequency is calculated using limiting dilution analysis software (e.g., ELDA) that applies Poisson statistics to determine the frequency of tumor-initiating cells in each population. Confidence intervals and statistical significance between different populations are determined using chi-square tests.
Emerging Therapeutic Approaches Targeting CSC Stemness
Strategic Approaches to CSC Eradication
The dual role of stemness in tumor initiation and maintenance presents corresponding dual opportunities for therapeutic intervention. Successful CSC-targeted therapies must address both the intrinsic properties of CSCs and their interactions with the tumor microenvironment.
Table 4: Therapeutic Strategies Targeting CSC Stemness Properties
| Therapeutic Strategy | Molecular Targets | Representative Agents | Development Status |
|---|---|---|---|
| Signaling Pathway Inhibition | Wnt, Notch, Hedgehog | Vismodegib, Demcizumab | Clinical trials |
| Differentiation Therapy | Retinoic acid receptors | All-trans retinoic acid (ATRA) | Approved for AML |
| Metabolic Targeting | OXPHOS, Glycolysis, Autophagy | Metformin, Chloroquine | Preclinical/Clinical trials |
| Immunotherapy Approaches | CSC-specific antigens | CAR-T, Bispecific antibodies | Early clinical trials |
| Microenvironment Disruption | CXCR4, IL-6, TGF-β | Plerixafor, Siltuximab | Clinical evaluation |
| Epigenetic Modulation | EZH2, BMI1, DNMTs | GSK126, Azacitidine | Preclinical/Clinical development |
Innovative CSC-Targeting Platforms
Recent advances in therapeutic platforms have enabled more precise targeting of CSC populations. Engineering stem cells themselves to generate renewable cancer-fighting immune cells represents a particularly innovative approach [20]. In a first-in-human clinical trial, UCLA scientists successfully reprogrammed a patient's blood-forming stem cells to generate a continuous supply of functional T cells targeting the NY-ESO-1 cancer marker [20]. This approach creates an "internal factory" that produces tumor-targeting immune cells over time, potentially offering longer-lasting protection against recurrence [20].
Bispecific antibody-drug conjugates represent another emerging platform for CSC targeting. Iza-bren, a bispecific ADC targeting EGFR and HER3 mutations, has shown promise in early clinical trials for non-small cell lung cancer, with 75% of patients receiving the optimal dose showing tumor regression [21]. This dual-targeting approach may help address the heterogeneity and adaptability of CSC populations.
Diagram 2: Multidimensional Therapeutic Strategies for Targeting CSCs. Effective CSC eradication requires combined approaches targeting stemness pathways directly, disrupting supportive niches, and forcing differentiation.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Research Reagents for CSC Investigation
| Reagent Category | Specific Products | Research Applications | Technical Considerations |
|---|---|---|---|
| CSC Surface Marker Antibodies | Anti-CD44, Anti-CD133, Anti-CD34, Anti-EPCAM, Anti-LGR5 | FACS isolation, Immunofluorescence, IHC | Species specificity, Clone validation, Multipanel compatibility |
| Stemness Transcription Factor Antibodies | Anti-OCT4, Anti-SOX2, Anti-NANOG, Anti-c-MYC | Intracellular staining, Western blot, ChIP | Fixation/permeabilization requirements, Nuclear localization |
| Pathway Modulators | Wnt agonists/antagonists, Notch inhibitors, Hh pathway blockers | Functional assays, Mechanism studies | Off-target effects, Dose optimization |
| Cell Culture Supplements | B27, N2, Recombinant EGF/FGF, KnockOut Serum Replacement | Tumor sphere assays, Clonal expansion | Batch-to-batch variability, Stability concerns |
| Proteomics Reagents | TMT/Isobaric tags, Phospho-enrichment materials, RPPA antibodies | Multi-omics profiling, Signaling analysis | Sample preparation rigor, Platform validation |
The dual role of stemness in tumor initiation and maintenance establishes CSCs as critical targets for oncogenic potential assessment and therapeutic development. Future research directions will need to address several key challenges, including the dynamic plasticity of CSC phenotypes, the lack of universal CSC biomarkers, and the complexities of CSC-microenvironment interactions [8]. Emerging technologies such as single-cell multi-omics, CRISPR-based functional screens, AI-driven analysis, and 3D organoid models are paving the way for more precise CSC characterization and targeting [8].
The clinical translation of CSC-targeting therapies will require careful assessment of therapeutic indices to avoid damaging normal tissue stem cells [22]. Additionally, the development of reliable biomarkers for monitoring CSC populations in patients during therapy remains an urgent need [16]. As our understanding of CSC biology deepens, integrative approaches combining metabolic reprogramming, immunomodulation, and targeted inhibition of core stemness pathways hold promise for overcoming therapy resistance and preventing tumor recurrence [8] [17]. The continued investigation of the dual role of stemness in cancer will undoubtedly yield crucial insights for both basic cancer biology and clinical oncology practice.
The study of mutation accumulation in stem cells is critical for understanding carcinogenesis and for assessing the oncogenic potential of stem cells intended for use in regenerative medicine. Somatic mutations acquired during a stem cell's lifetime can initiate tumorigenesis, making the rate and patterns of this accumulation a key area of research. This guide provides a direct comparison of mutation rates between developmental and adult stages of stem cells, synthesizing quantitative data, experimental methodologies, and molecular mechanisms to inform researchers and drug development professionals.
Quantitative Comparison of Mutation Rates
Data from genomic studies reveal distinct patterns of mutation accumulation in stem cells during developmental and adult life stages. The tables below summarize key quantitative findings.
Table 1: Comparison of Developmental and Adult Stem Cell Mutation Rates
| Life Stage | Stem Cell Type | Mutation Rate | Key Influencing Factors |
|---|---|---|---|
| Developmental | Omnipotent stem cells (immediately after conception) | 2–3 mutations per division [23] | Pre-genome activation, limited DNA repair, diluted maternal factors [23] |
| Developmental | Stem cells (between conception and gastrulation) | ~1.6 mutations per division [23] | Rapid proliferation, chromatin remodeling, lenient DNA damage checkpoints [23] |
| Developmental | Hematopoietic Stem/Progenitor Cells (HSPCs) | 5.8-fold higher per year than postnatal rate [23] | Developmental processes and proliferation rate |
| Adult | Pluripotent Stem Cells (PSCs) in vitro | 3.5 ± 0.5 base substitutions per population doubling [24] | Culture conditions, particularly oxidative stress at 20% O₂ [24] |
| Adult | Intestinal & Liver Adult Stem Cells (ASCs) in vitro | 7.2 ± 1.1 and 8.3 ± 3.6 base substitutions per population doubling, respectively [24] | Culture conditions, oxidative stress [24] |
| Adult | Intestinal, Colon, & Liver ASCs in vivo | ~40 novel mutations per genome per year [25] | Endogenous mutational processes, tissue-specific cell division rates [25] |
Table 2: Impact of Culture Conditions on PSC Mutation Rates
| Culture Condition | Mutation Rate | Effect Compared to Standard Conditions |
|---|---|---|
| Standard (20% O₂) | 0.28-0.37 x 10⁻⁹ SNVs per base-pair per day [26] | Baseline rate [26] |
| Low Oxygen (5% O₂) | 0.13 x 10⁻⁹ SNVs per base-pair per day [26] | >50% reduction in mutation rate [26] |
| Low Oxygen (3% O₂) | Not explicitly quantified | Lower mutational load, specifically in mutations linked to oxidative stress [24] |
| With ROCK Inhibitor (Y27632) | Not significantly different from standard [26] | No substantial impact on mutation rate [26] |
Experimental Protocols for Mutation Detection
Accurately measuring the low burden of somatic mutations in normal stem cells requires sophisticated, high-fidelity techniques.
Clonal Culture and Whole-Genome Sequencing
This method involves expanding a single stem cell into a clonal population to obtain sufficient DNA for whole-genome sequencing (WGS). Any mutation present in the original founding cell will be present in all descendant cells and thus show a high variant allele frequency (VAF) in the sequencing data. To measure mutations accumulated during a specific period, a subcloning step is performed after a defined culture period. Mutations found in the subclone, but not the original clone, represent those acquired during the intervening time [24] [23]. This approach effectively filters out germline variants and allows for the calculation of mutation rates per population doubling.
Duplex Sequencing (NanoSeq)
Duplex sequencing is an error-corrected bulk sequencing method that provides single-molecule sensitivity. The latest NanoSeq protocols use optimized fragmentation (sonication or enzymatic) and dideoxynucleotides during library preparation to prevent error transfer between DNA strands, achieving an ultralow error rate of below 5 errors per billion base pairs. This allows for the accurate detection of somatic mutations present in single cells within a polyclonal sample, without the need for clonal expansion. It is particularly powerful for profiling the driver mutation landscape across hundreds of clones from a single sample [27].
Signaling Pathways and Mutational Processes
The mutational processes active in stem cells leave distinct footprints in the genome, known as mutational signatures.
Oxidative Stress as a Key Driver in Vitro
A dominant mutational process in standard stem cell cultures is oxidative stress. Culturing pluripotent and adult stem cells under atmospheric oxygen (20% O₂) leads to a mutation spectrum dominated by C>A transversions, a signature linked to reactive oxygen species (ROS) [24]. This signature is significantly reduced when cells are cultured under physiological oxygen tension (3-5% O₂), demonstrating that oxidative stress is a major, modifiable contributor to in vitro mutation accumulation [26] [24].
Developmental vs. Adult Mutational Processes
The high mutation rate in early development is attributed to a unique biological context: cell division occurs before the full activation of the genome and the associated transcription-coupled repair machinery, and maternal DNA repair factors become diluted. Additionally, rapid proliferation, extensive chromatin remodeling, and more lenient cell-cycle checkpoints contribute to unavoidable mutation accumulation during this critical period [23].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for Studying Stem Cell Mutations
| Reagent / Material | Function in Experimental Protocol |
|---|---|
| Rho Kinase (ROCK) Inhibitor (Y27632) | Improves survival of human pluripotent stem cells (PSCs) after passaging, though it does not significantly alter the underlying mutation rate [26]. |
| Clinical Grade Human ES/iPS Cell Lines | Provides a standardized, physiologically relevant model for quantifying mutation rates in a therapeutic context [26] [28]. |
| Organoid Culture Systems | Enables the long-term, clonal expansion of primary adult stem cells (e.g., from intestine, liver) for mutation accumulation studies [24] [25]. |
| Duplex Sequencing (NanoSeq) Reagents | Ultra-low error rate sequencing is essential for detecting very low-frequency somatic mutations in polyclonal samples or single DNA molecules [27]. |
| Targeted Sequencing Panels | Focused gene panels allow for cost-effective, deep sequencing to profile driver mutation landscapes across hundreds of samples [27]. |
Stem cell mutation rates are not static, but are highly dependent on the biological context, with developmental stages exhibiting higher per-division rates than most adult tissues. A critical finding for regenerative medicine is that standard in vitro culture conditions can dramatically increase the mutational load in stem cells, primarily through oxidative stress. However, this risk is modifiable, as culturing under physiologically low oxygen tension significantly reduces mutation accumulation. These insights are paramount for developing safer stem cell-based therapies and for accurately assessing their oncogenic potential. Future research leveraging even more sensitive sequencing technologies will continue to refine our understanding of the complex interplay between mutation, selection, and cancer initiation in stem cell pools.
The stem cell niche is a dynamic microenvironment that plays a critical role in regulating stem cell behavior, maintenance, and transformation. Comprising various cellular components, signaling molecules, and physical factors, the niche provides essential cues that balance self-renewal and differentiation. Disruption of this delicate balance can drive malignant transformation, leading to cancer initiation and progression. This review systematically compares the oncogenic potential across different stem cell types by examining their unique niche interactions, supported by experimental data and signaling pathway analysis. Understanding these mechanisms provides crucial insights for developing targeted cancer therapies and assessing transformation risks in regenerative medicine applications.
The concept of the stem cell niche was first formally proposed by R. Schofield in 1978, hypothesizing that specialized microenvironments within tissues preserve stem cell proliferative potential and block maturation [29]. This niche hypothesis has since been validated across numerous mammalian tissues, with the niche functioning as a fundamental regulatory unit that governs stem cell fate decisions through direct cell-cell contact, secreted factors, and physical constraints [29] [30]. The niche maintains tissue homeostasis by precisely balancing stem cell activity—ensuring adequate self-renewal while producing differentiated progeny for tissue maintenance and repair.
When normal niche function is disrupted, the resulting dysregulation of stem cell behavior can initiate transformation processes. Cancer stem cells (CSCs), a subpopulation within tumors that exhibit stem-like properties, similarly depend on specialized "CSC niches" for their maintenance and protection [31]. These CSC niches share functional similarities with normal stem cell niches but promote tumorigenesis, immune evasion, and therapy resistance through aberrant signaling [32] [31]. This comparison guide examines how niche interactions influence transformation potential across stem cell types, providing experimental methodologies and analytical frameworks for oncogenic risk assessment.
Comparative Analysis of Stem Cell Niches and Transformation Risks
Table 1: Stem Cell Niche Characteristics and Transformation Potential Across Tissue Types
| Stem Cell Type | Key Niche Support Cells/Components | Critical Signaling Pathways | Transformation Risks/Cancer Associations | Experimental Model Systems |
|---|---|---|---|---|
| Hematopoietic Stem Cells (HSCs) | Osteoblasts, vascular cells, perivascular cells [29] | CXCL12, Wnt, Notch, ANG1 [29] | Acute Myeloid Leukemia (CD34+/CD38- phenotype) [32] [3] | Serial transplantation in immunodeficient mice [29] [8] |
| Intestinal Stem Cells | Paneth cells, vascular cells, fibroblasts [29] | Wnt, BMP, Notch [29] | Colorectal cancer (LGR5+ or CD133+ CBC cells) [29] [8] | Lineage tracing with Lgr5-CreERT2; intestinal organoids [29] |
| Neural Stem Cells | Vascular cells, ependymal cells, astrocytes [29] | Wnt, SHH, FGF, VEGF, Notch [29] | Glioblastoma (CD133+ or Nestin+ cells) [8] [3] | In vitro neurosphere assays; patient-derived organoids [8] |
| Mesenchymal Stem Cells (MSCs) | Bone marrow stromal cells, adipocytes [33] | TGF-β, PDGFR-β/GPR91 [32] [33] | Sarcoma; niche-induced transformation via metabolic symbiosis [32] [7] | In vivo tracking with PET/MRI; immunocompromised mouse models [7] |
| Embryonic Stem Cells (ESCs) | Trophoblast cells, stromal fibroblasts [7] | LIF/STAT3, TGF-β/Activin [7] | Teratoma formation (testicular teratomas in 129-strain mice) [8] [3] | Teratoma assay in immunodeficient mice [7] |
Table 2: Quantitative Assessment of Stem Cell Transformation Potential
| Stem Cell Type | Transplantation Tumorigenicity Threshold | Time to Tumor Formation | CSC Frequency in Resulting Tumors | Key Transformation Markers |
|---|---|---|---|---|
| HSCs (AML) | 1,000-10,000 CD34+/CD38- cells [3] | 8-12 weeks [3] | 0.2-1% in primary AML [3] | CD34+/CD38-, ALDH1 activity [32] [3] |
| Intestinal Stem Cells | As few as 200 LGR5+ cells [29] | 4-8 weeks [29] | 0.4-20% (varies by cancer stage) [8] [3] | LGR5, CD133, CD44 [29] [8] |
| Breast Stem Cells | 200 CD44+CD24-/low cells [3] | 12 weeks [3] | 3-4x higher in stage III vs. stage I [3] | CD44+/CD24-/low, ALDH1+ [32] [3] |
| Glioblastoma Stem Cells | 1,000-10,000 CD133+ cells [3] | 10-16 weeks [3] | 1-20% (depending on tumor subtype) [3] | CD133, Nestin, SOX2 [8] [3] |
| Melanoma Stem Cells | Varies significantly by model [8] | 6-20 weeks [8] | 0.4-82.5% (highly variable) [3] | CD133, CD166, ABC transporters [8] |
Experimental Protocols for Assessing Niche-Driven Transformation
Lineage Tracing and Fate Mapping
Lineage tracing represents a fundamental methodology for identifying stem cell populations and characterizing their niche interactions in vivo [29]. The protocol utilizes genetic labeling to mark specific cell populations and track their progeny over time, enabling researchers to visualize differentiation hierarchies and identify cells with multilineage differentiation potential—a hallmark of stemness.
Detailed Protocol:
- Genetic Marker Selection: Choose a stem cell-specific promoter (e.g., Lgr5 for intestinal stem cells, Nestin for neural stem cells) to drive Cre recombinase expression [29]
- Reporter System: Cross with reporter mice containing a loxP-flanked STOP cassette preceding a fluorescent protein (e.g., tdTomato, GFP)
- Induction: Administer tamoxifen to activate CreERT2, inducing recombination and permanent label expression in target cells and their descendants
- Temporal Analysis: Track labeled cells at multiple timepoints (days to months) to assess self-renewal and differentiation capacity
- Tissue Processing: Analyze tissue sections via immunohistochemistry and fluorescence microscopy to quantify lineage contributions
This approach demonstrated that Lgr5+ crypt base columnar cells function as intestinal stem cells and can give rise to intestinal adenomas upon Apc deletion, identifying them as a cell-of-origin for colorectal cancer [29].
Serial Transplantation Assays
The gold standard for assessing functional stem cell activity, particularly for hematopoietic stem cells, involves serial transplantation into immunocompromised recipient animals [29] [8]. This method quantitatively measures self-renewal capacity—a critical property of both normal stem cells and CSCs.
Detailed Protocol:
- Cell Isolation: Purify candidate stem cell populations using fluorescence-activated cell sorting (FACS) with specific surface markers (e.g., CD34+CD38- for leukemia, CD44+CD24- for breast cancer)
- Primary Transplantation: Inject limiting dilutions of test cells into primary immunodeficient recipients (NOD/SCID or NSG mice)
- Engraftment Assessment: Monitor recipients for tumor development or tissue reconstitution over 8-24 weeks
- Secondary Transplantation: Isolate cells from primary recipients and transplant into secondary recipients to assess self-renewal capacity
- Quantitative Analysis: Calculate stem cell frequency using extreme limiting dilution analysis (ELDA) software
This methodology identified CD34+CD38- cells as acute myeloid leukemia stem cells, demonstrating their ability to recapitulate disease in recipient mice while more differentiated cells lacked this potential [8] [3].
Single-Cell Multi-Omics Analysis
Advanced sequencing technologies enable comprehensive characterization of stem cell heterogeneity and niche interactions at single-cell resolution, providing insights into transformation mechanisms [32] [8].
Detailed Protocol:
- Single-Cell Suspension: Dissociate tissue or tumor samples into single-cell suspensions while maintaining viability
- Cell Partitioning: Load cells into droplet-based systems (10X Genomics) for barcoding
- Library Preparation: Generate transcriptome (scRNA-seq), epigenome (scATAC-seq), or multimodal libraries
- Sequencing: Perform high-throughput sequencing on Illumina platforms
- Bioinformatic Analysis:
- Cluster cells based on expression profiles using Seurat or Scanpy
- Reconstruct differentiation trajectories with pseudotime algorithms (Monocle, PAGA)
- Identify cell-cell communication networks (NicheNet, CellChat)
- Correlate transcriptional states with niche localization through spatial transcriptomics
This approach has revealed the plastic nature of CSCs and their adaptive responses to microenvironmental cues, explaining how non-CSCs can reacquire stem-like properties under therapeutic pressure [8].
Signaling Pathways in Niche-Mediated Transformation
Niche-Activated Signaling Pathways in Stem Cell Transformation
The niche regulates stem cell behavior through complex signaling networks that, when dysregulated, drive oncogenic transformation. The diagram above illustrates key pathways implicated in this process, with the following mechanistic insights:
Wnt/β-Catenin Pathway: Niche-derived Wnt ligands maintain stemness in intestinal crypts and hematopoietic systems [29]. Dysregulation through APC mutations or β-catenin stabilization transforms LGR5+ intestinal stem cells, initiating colon carcinoma within days [29]. In hepatocellular carcinoma, β-catenin forms a positive feedback loop with PD-L1, simultaneously promoting immune evasion and stemness [31].
Notch Signaling: Mediates direct cell-cell communication between stem cells and niche cells [29]. In intestinal and neural niches, Notch activation inhibits differentiation, maintaining stem cell pools. In breast and pancreatic cancers, aberrant Notch activation expands CSC populations and drives therapy resistance [3].
Hedgehog Signaling: Regulates stem cell quiescence in multiple tissues [3]. In basal cell carcinoma and medulloblastoma, mutational activation of Hedgehog signaling drives tumor initiation from stem cells [3]. ADAR1-mediated RNA editing of GLI1 in hepatic CSCs enhances nuclear localization, promoting tumor initiation [32].
TGF-β/BMP Pathway: Exhibits context-dependent effects, often suppressing stemness in normal tissues but promoting epithelial-mesenchymal transition (EMT) and CSC phenotypes in advanced cancers [3]. In head and neck squamous cell carcinoma, TGF-β induces CD80 expression on CSCs, enabling immune evasion [31].
JAK/STAT Signaling: Regulates stem cell maintenance in Drosophila testes and mammalian epithelia [29]. Persistent STAT3 activation in CSCs promotes self-renewal and upregulates PD-L1 expression, contributing to immunosuppression [31].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Niche-Driven Transformation
| Reagent/Category | Specific Examples | Research Application | Transformation Assessment Utility |
|---|---|---|---|
| Cell Surface Markers | CD34, CD38, CD44, CD24, CD133, LGR5, EpCAM | FACS isolation of stem cell populations | Identifies and purifies CSCs for functional assays [8] [3] |
| Signaling Pathway Inhibitors | DKK1 (Wnt inhibitor), DAPT (Notch inhibitor), Cyclopamine (Hedgehog inhibitor), SD-208 (TGF-β inhibitor) | Pathway perturbation studies | Tests functional dependency of CSCs on specific niche signals [3] |
| Cytokines/Growth Factors | CXCL12, EGF, FGF, TGF-β, BMP4, Wnt3a | In vitro niche reconstitution | Maintains stemness in culture and supports organoid formation [29] |
| Reporter Systems | Lgr5-GFP, Axin2-lacZ, PD-L1-GFP, TCF/LEF-GFP | Lineage tracing and signaling activity | Visualizes stem cell locations and pathway activation in real-time [29] |
| Immune Checkpoint Reagents | Anti-PD-1/PD-L1, anti-CTLA-4, anti-CD47, anti-CD24 | Immune evasion studies | Evaluates CSC ability to evade immune surveillance [31] |
| Metabolic Probes | 2-NBDG (glucose uptake), MitoTracker, C11-BODIPY581/591 (lipid peroxidation) | Metabolic profiling | Assesses metabolic adaptations in CSCs within hypoxic niches [32] [8] |
Discussion: Clinical Implications and Therapeutic Targeting
The comparative analysis reveals that transformation risk varies significantly across stem cell types, influenced by their respective niche interactions, turnover rates, and intrinsic regulatory mechanisms. Hematopoietic and intestinal stem cells demonstrate high susceptibility to niche-driven transformation, correlating with their high proliferative activity and well-defined hierarchical organizations [29] [8]. In contrast, mesenchymal stem cells exhibit lower inherent transformation risk but can promote tumor progression through paracrine signaling within established tumor microenvironments [7] [33].
Emerging therapeutic strategies aim to target niche interactions to eliminate CSCs while sparing normal stem cells. Approaches include:
- Niche Disruption: Targeting CSC-TME interactions through CXCR4 inhibitors to disrupt protective niches in leukemia [32]
- Immune Evasion Blockade: Combining PD-1/PD-L1 inhibitors with CSC-specific vaccines to overcome immune privilege [32] [31]
- Metabolic Interdiction: Dual targeting of glycolysis and oxidative phosphorylation to exploit CSC metabolic dependencies [8]
- Differentiation Therapy: Inducing CSC differentiation through BMP signaling to reduce therapy resistance [3]
Advanced model systems including 3D organoids, CRISPR-based functional screens, and AI-driven multiomics integration are accelerating the development of niche-targeted therapies [8]. Future research should focus on resolving niche heterogeneity at single-cell resolution across tumor types and developmental stages to identify context-specific vulnerabilities for therapeutic exploitation.
Assessment Arsenal: From Traditional Models to Cutting-Edge Screening Platforms
In the field of stem cell research and therapeutic development, assessing oncogenic potential represents a critical safety imperative. Tumorigenicity testing in immunocompromised mice serves as the gold-standard methodology for evaluating the risk of unwanted cell growth in living organisms, forming an essential component of the safety assessment for novel cell-based therapies [7]. These specialized animal models enable researchers to study the behavior of human-derived cells in an in vivo environment while circumventing the immune-mediated rejection that would normally eliminate foreign cells, thereby allowing for the detection of malignant transformation and uncontrolled proliferation [34].
The fundamental principle underlying these models involves implanting human stem cells or their differentiated derivatives into mice with compromised immune systems and monitoring for tumor formation over time. This approach provides invaluable preclinical data on the safety profile of cellular products, particularly for pluripotent stem cell-derived therapies where residual undifferentiated cells could pose significant tumorigenic risks [7]. As regenerative medicine continues to advance toward clinical applications, robust tumorigenicity assessment using these models has become indispensable for regulatory approval and patient safety, helping to identify and mitigate the risks of teratoma formation and malignant transformation before human trials commence [7].
Comparative Analysis of Immunocompromised Mouse Models
The evolution of immunocompromised mouse models has progressed significantly since the first nude mice were discovered in the 1960s, with each successive generation offering improved engraftment rates and broader applications for tumorigenicity testing [35] [34]. The selection of an appropriate model depends on multiple factors including the cell type being tested, required observation period, and specific research questions. The table below provides a comprehensive comparison of the most commonly utilized immunocompromised mouse strains in tumorigenicity assessment.
Table 1: Comparison of Immunocompromised Mouse Models for Tumorigenicity Testing
| Mouse Strain | Genetic Mutation(s) | Immune Deficiencies | Success Rate for PDX | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Nude mice | Foxn1 | T-cell deficient | Low | Easy tumor monitoring; Cost-effective | Functional B cells and NK cells; Lower engraftment success |
| SCID mice | Prkdc | T-cell and B-cell deficient | Low | Better implantation than nude mice | NK cell activity; Radio-sensitive; Lymphocyte "leakage" |
| NOD-SCID mice | SCID (Prkdc) + NOD background | T-cells, B-cells, reduced NK cells, defective macrophages/dendritic cells | Moderate | Improved engraftment over SCID | Spontaneous lymphoma; Short lifespan; Radio-sensitive |
| NOG/NSG/NOJ mice | SCID + IL2Rγ null (Jak3) | T-cells, B-cells, NK cells, reduced macrophage/dendritic function | High | Outstanding engraftment success; Longer lifespan | Require strict SPF conditions; Higher cost |
| BRG/BRJ mice | IL2Rγ, Jak3, Rag-2 | T-cells, B-cells, NK cells, functional macrophages | High | Excellent engraftment; Radiation-resistant | Higher cost; Limited availability |
The progression from nude to more severely immunocompromised strains like NSG (NOD-scid gamma) mice has dramatically improved the success rates of patient-derived xenograft (PDX) models, which are crucial for accurate tumorigenicity assessment [34]. The NSG strain, with its deficiencies in T cells, B cells, and natural killer (NK) cells, along with defective macrophage and dendritic cell function, provides the most permissive environment for engrafting human cells, including those with lower tumorigenic potential [34]. This high level of immunodeficiency makes NSG mice particularly valuable for detecting minimal residual tumorigenic cells in stem cell populations, a critical consideration for clinical safety.
Experimental Design and Methodological Framework
Model Establishment and Cell Implantation
The construction of robust tumorigenicity models requires careful consideration of implantation methods and locations. Two primary approaches dominate the field: subcutaneous injection and orthotopic implantation. Subcutaneous injection involves depositing cells into the loose connective tissue beneath the skin, allowing for direct visual monitoring of tumor growth through simple caliper measurements [34]. This method offers technical simplicity and straightforward monitoring but may lack the appropriate tissue microenvironment that influences tumor development. Alternatively, orthotopic implantation places cells into the equivalent tissue of origin in the mouse (e.g., neural stem cells into brain tissue), potentially providing a more biologically relevant microenvironment but requiring more sophisticated monitoring techniques such as in vivo imaging [36].
The preparation of cells for implantation typically follows one of two strategies: single-cell suspensions or tissue fragment implantation. Single-cell suspensions, often prepared using enzymatic digestion, provide more controlled cell dosing and reduce sample heterogeneity, but may compromise cellular viability and intercellular interactions through the dissociation process [34]. Conversely, tissue fragment implantation better preserves native tissue architecture and cell-cell interactions, potentially maintaining crucial survival signals, though with less precise control over the number of cells implanted [34]. For tumorigenicity testing of stem cell populations, the use of basement membrane matrix (such as Matrigel) as a carrier has demonstrated improved engraftment efficiency by providing structural support and essential survival signals [34].
Monitoring and Endpoint Analysis
Comprehensive tumorigenicity assessment requires multimodal monitoring throughout the experimental timeline. The standard observation period typically extends from several weeks to months, with specific duration determined by the expected doubling time of the tested cells and their theoretical tumorigenic potential [7]. Regular tumor volume measurements using calipers (for subcutaneous models) or advanced imaging techniques (for orthotopic models) provide growth kinetics data. In vivo imaging technologies, including bioluminescent and fluorescent reporters, enable longitudinal tracking of cell survival and proliferation without sacrificing animals at intermediate timepoints [35].
Endpoint analyses encompass both gross pathological examination and detailed histological assessment. Histological staining of explanted tissues remains the definitive method for identifying tumor formation and characterizing tumor type [7]. Key techniques include hematoxylin and eosin (H&E) staining for general tissue architecture and identification of undifferentiated cells, immunohistochemistry for specific markers of pluripotency (OCT4, NANOG, SOX2) or differentiation, and special stains to identify tissue types representative of all three germ layers in the case of teratoma formation [7]. Additional molecular analyses may include PCR-based detection of human-specific genes to confirm human cell origin and quantify biodistribution beyond the implantation site [7].
Table 2: Key Analytical Methods in Tumorigenicity Assessment
| Method Category | Specific Techniques | Primary Application | Key Output Parameters |
|---|---|---|---|
| In Vivo Monitoring | Caliper measurements, Bioluminescent imaging, PET/CT | Tumor growth tracking | Tumor volume, Growth kinetics, Cell survival |
| Histopathological Analysis | H&E staining, Immunohistochemistry, Special stains | Tumor identification and characterization | Tissue architecture, Pluripotency markers, Teratoma composition |
| Molecular Analysis | qPCR, Biodistribution studies, Genomic integration assays | Origin and distribution assessment | Human cell quantification, Off-target engraftment, Genetic stability |
| Functional Assessment | Secondary transplantation, Differentiation assays | Malignant potential evaluation | Self-renewal capacity, Differentiation potential |
Research Reagent Solutions for Tumorigenicity Testing
The successful implementation of tumorigenicity assays requires access to specialized reagents and resources. The following table outlines essential research tools and their applications in model development and analysis.
Table 3: Essential Research Reagents and Resources for Tumorigenicity Testing
| Reagent/Resource | Function | Application Examples |
|---|---|---|
| Basement Membrane Matrix (Matrigel) | Provides extracellular matrix support for engrafted cells | Enhancing cell survival and engraftment efficiency after implantation [34] |
| Immunodeficient Mouse Strains | Host environment for human cell engraftment | NSG, NOG, NOD-SCID strains for tumorigenicity studies [34] |
| Cell Line Tracking Systems | Longitudinal monitoring of cell fate | Bioluminescent (luciferase) or fluorescent (GFP) reporters for in vivo imaging [35] |
| PDX Biobanks | Source of validated tumor models | NCI PDMR, EurOPDX Consortium, Jackson Laboratory PDX resources [34] |
| Human-Specific Detection Assays | Discrimination of human vs. mouse cells | qPCR for human-specific Alu sequences, human MHC immunohistochemistry [7] |
| Pluripotency Markers | Identification of undifferentiated stem cells | Antibodies against OCT4, NANOG, SOX2, SSEA-4 for immunohistochemistry [7] |
Experimental Workflow for Tumorigenicity Testing
The following diagram illustrates the standard workflow for conducting tumorigenicity testing in immunocompromised mice, from experimental design through data interpretation:
Integration with Complementary Preclinical Models
While immunocompromised mouse models represent the current gold standard for tumorigenicity testing, they are most informative when integrated with complementary preclinical approaches. Advanced in vitro systems such as 3D organoid cultures and organ-on-a-chip platforms provide valuable preliminary safety data with higher throughput capabilities [37]. These systems can model aspects of the tumor microenvironment and cell-cell interactions that influence tumorigenic potential, serving as important screening tools before progressing to more resource-intensive in vivo studies [37].
The emergence of proteomic-based stemness indices (PROTsi) and other computational approaches offers promising avenues for predicting tumorigenic risk based on molecular signatures [14]. These tools can quantify oncogenic dedifferentiation in relation to histopathological features and molecular profiles, potentially identifying high-risk cell populations before in vivo implantation [14]. Similarly, innovative imaging technologies such as immuno-PET with targeted tracers enable non-invasive monitoring of specific cell populations and their functional status in living animals, providing dynamic information about engrafted cell behavior [38].
Tumorigenicity testing in immunocompromised mice remains an indispensable component of the safety assessment pipeline for stem cell-based therapies. The strategic selection of appropriate mouse models, coupled with robust experimental design and comprehensive endpoint analyses, provides critical data on oncogenic risk that informs clinical translation decisions. As the field advances, the integration of these gold-standard in vivo models with emerging technologies in molecular imaging, multi-omics analysis, and complex in vitro systems will further enhance our ability to predict and mitigate tumorigenic potential in regenerative medicine applications.
Researchers must carefully match model selection to their specific research questions, considering the balance between engraftment efficiency, physiological relevance, and practical constraints. The continued refinement of these models and the development of standardized assessment protocols will strengthen the preclinical safety evaluation process, ultimately supporting the development of safer cell-based therapies for patients with unmet medical needs.
The assessment of oncogenic potential—the ability of cells to form tumors—is a critical step in stem cell research, safety profiling for cell therapies, and cancer biology. For decades, the soft agar colony formation assay has been the gold-standard in vitro method for detecting anchorage-independent growth, a hallmark of cellular transformation. However, the rise of more physiologically relevant three-dimensional (3D) models, particularly organoids, is revolutionizing the field. This guide provides a detailed, objective comparison of these two pivotal techniques, framing them within the context of modern oncogenic risk assessment across different stem cell types. While the soft agar assay offers a straightforward, quantitative readout of transformation, 3D organoid cultures deliver unprecedented physiological relevance, recapitulating the complex architecture and cell-cell interactions of in vivo tissues [39] [40]. Understanding the strengths, limitations, and appropriate applications of each method is essential for researchers and drug development professionals aiming to accurately evaluate the tumorigenic risk of novel stem cell-based therapies or to model cancer initiation and progression.
Fundamental Technique Comparison
The following table summarizes the core characteristics, advantages, and limitations of the soft agar and 3D organoid methods.
Table 1: Core Characteristics of Soft Agar and 3D Organoid Assays
| Feature | Soft Agar Colony Formation Assay | 3D Organoid Cultures |
|---|---|---|
| Core Principle | Measures anchorage-independent growth in a semi-solid medium [41]. | Stem cells self-organize into 3D structures mimicking organ architecture [39] [42]. |
| Key Readout | Number and/or size of colonies formed after 2-4 weeks [41]. | Organoid morphology, growth, viability, and differentiation over long-term culture [39] [40]. |
| Throughput | High; suitable for drug screening and large-scale experiments [41]. | Variable; can be medium to high with specialized platforms, but often lower than soft agar [40] [43]. |
| Physiological Relevance | Low; lacks tissue context, ECM, and complex cell interactions [40]. | High; recapitulates cell-ECM interactions, nutrient gradients, and in vivo-like architecture [39] [40]. |
| Key Applications | - Identifying transformed cells- Oncogene validation- Drug cytotoxicity screening [41] | - Disease modeling (including cancer)- Drug screening & personalized medicine- Host-microbe interaction studies [39] [44] |
| Primary Limitations | - Does not model tissue-specific contexts- Can yield false positives/negatives- Opaque matrix complicates imaging [41] | - Technically challenging & costly- Batch-to-batch variability (e.g., Matrigel)- Limited scalability & standardization [40] [43] |
Application in Oncogenic Potential Assessment
When specifically applied to evaluating the oncogenic risk of different stem cell types, the two assays provide complementary insights. The choice of model depends heavily on the research question, balancing throughput with biological complexity.
Table 2: Assessing Oncogenic Potential Across Stem Cell Types
| Stem Cell Type | Oncogenic Risk & Context | Soft Agar Assay Utility | 3D Organoid Model Utility |
|---|---|---|---|
| Induced Pluripotent Stem Cells (iPSCs) | High risk due to reprogramming factors and culture-induced mutations; critical for safety of regenerative therapies [7]. | Excellent for initial, high-throughput screening of transformation events in undifferentiated or differentiated iPSCs. | Models multi-stage transformation within a tissue context; can co-culture with stromal cells to assess microenvironmental impact [39]. |
| Mesenchymal Stem/Stromal Cells (MSCs) | Lower but present risk; potential for transformation after long-term culture; used in numerous clinical trials [7] [45]. | Standardized method for quality control during biomanufacturing to ensure safety prior to clinical application. | Patient-derived tumor organoids can be used to study MSC-tumor interactions and assess pro- or anti-tumorigenic effects of MSC therapies [8]. |
| Cancer Stem Cells (CSCs) | Defined by their tumor-initiating and therapy-resistant capabilities; drivers of metastasis and relapse [8]. | Classic assay to demonstrate the anchorage-independent growth of isolated CSCs and test CSC-targeting drugs. | Highly relevant; Tumor organoids derived from patients preserve CSC hierarchy and heterogeneity, enabling study of drug resistance and recurrence [42] [8]. |
| Embryonic Stem Cells (ESCs) | Risk of teratoma formation upon transplantation; a major barrier to clinical translation [7]. | Used to test the transformative potential of differentiated ESCs before in vivo use. | Brain, intestinal, and other organoids model tissue-specific cancers, revealing how mutations in developmental pathways drive transformation [39]. |
Soft Agar Colony Formation Protocol The standard protocol involves creating a two-layer system in a multi-well plate. A base layer of 0.5% - 1.0% agar in culture medium is solidified to prevent cell attachment to the plastic. A top layer, consisting of 0.3% - 0.4% agar in medium containing the single-cell suspension of interest (e.g., stem cells), is then poured over the base layer. The low-concentration agar creates a semi-solid mesh that allows nutrient diffusion but prevents anchorage. Cells are cultured for 2-4 weeks, with media refreshed periodically. Colonies are then stained with dyes like iodonitrotetrazolium chloride (INT) or MTT and counted manually or with an automated colony counter [41]. Variants like the methylcellulose clonogenic assay operate on a similar principle of preventing adhesion [42].
3D Organoid Culture for Cancer Modeling Protocols for generating tumor organoids are more diverse and complex. In one common method for colorectal cancer (CRC) research:
- Tissue Digestion: Patient-derived tumor tissue or PDX tissue is minced and enzymatically digested into small fragments or single cells.
- Matrix Embedding: The cell suspension is mixed with a basement membrane extract (e.g., Matrigel) and plated as droplets in a multi-well plate. The plate is incubated to allow the matrix to polymerize.
- Specialized Media: The embedded culture is overlaid with a specific organoid growth medium containing a cocktail of growth factors and inhibitors (e.g., Wnt agonists, R-spondin, Noggin) tailored to maintain the cancer stem cell niche [42] [40].
- Culture and Passaging: Organoids grow and are monitored for formation of complex, cystic, or solid structures. They can be passaged every 1-4 weeks by mechanically breaking and enzymatically digesting the organoids, followed by re-embedding in fresh matrix [42] [40]. Recent studies have optimized these methods across eight CRC cell lines, demonstrating how co-culture with fibroblasts in a 3D setting can enhance physiological relevance for drug screening [40].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these assays requires specific, high-quality reagents. The following table details essential materials and their functions.
Table 3: Key Reagent Solutions for Featured Assays
| Reagent / Material | Function and Application | Key Considerations |
|---|---|---|
| Agarose / Agar | Forms the semi-solid matrix for the soft agar assay, preventing cell adhesion and enabling quantification of anchorage-independent growth [41]. | Purity and gelling temperature are critical. Low-melting-point agarose is often preferred for the cell-containing top layer. |
| Basement Membrane Extract (e.g., Matrigel) | A natural scaffold derived from mouse sarcoma, used to provide a physiological 3D environment for organoid growth and differentiation [39] [42] [40]. | High batch-to-batch variability; undefined composition; requires cold handling. A major focus is developing synthetic alternatives [43]. |
| Defined Organoid Media Kits | Tailored formulations of growth factors, hormones, and small molecules (e.g., Wnt, R-spondin, Noggin) that support the self-renewal and differentiation of stem cells into specific organoid types [42] [43]. | Essential for long-term culture and biobanking. Compositions are often proprietary and organ-specific. |
| Low-Attachment Plates | Surface-treated plastic plates that minimize cell adhesion, used for scaffold-free spheroid generation and some organoid cultures via the liquid overlay technique [39] [40]. | A cost-effective alternative to matrix-embedding for some applications. Critical for generating tumorspheres from CSCs [42]. |
| LA717 (Low Molecular Weight Agar) | A novel additive that disperses cells in liquid culture, preventing aggregate fusion and enabling formation of uniform, clonal spheroids suitable for high-content imaging and drug screening [41]. | Keeps the medium clear with minimal added viscosity, overcoming key limitations of traditional soft agar and other 3D matrices [41]. |
Visualizing Pathways and Workflows
Cancer Stem Cell Signaling in Organoid Transformation
This diagram illustrates key signaling pathways that are often dysregulated in cancer stem cells (CSCs) and can be studied within 3D organoid models to assess oncogenic potential.
Diagram Title: Key Signaling Pathways Driving CSC Oncogenic Potential
Experimental Workflow for Oncogenic Risk Assessment
This workflow outlines a parallel experimental strategy using both soft agar and organoid models to thoroughly evaluate the oncogenic potential of stem cell populations.
Diagram Title: Parallel Workflows for Oncogenic Risk Assessment
The choice between soft agar colony formation assays and 3D organoid cultures is not a matter of declaring a single superior technology, but of selecting the right tool for the specific research question. The soft agar assay remains a powerful, high-throughput workhorse for the direct and quantitative assessment of anchorage-independent growth, making it indispensable for initial safety screening and transformation studies. In contrast, 3D organoid cultures offer a transformative level of biological fidelity, enabling researchers to probe oncogenic potential within a tissue-like context that captures stem cell hierarchy, heterogeneity, and microenvironmental interactions. For a comprehensive oncogenic risk assessment profile, particularly for novel stem cell therapies, a sequential or parallel approach using both methods can provide robust data: the quantitative power of soft agar complemented by the physiological insights of organoids. As organoid technology continues to advance through improved standardization, vascularization, and the development of defined synthetic matrices, its role in predicting clinical outcomes and ensuring the safety of regenerative medicines is poised to expand dramatically [43].
The functional characterization of cancer stem cells (CSCs) represents a critical frontier in oncology research, particularly for understanding tumor initiation, therapeutic resistance, and metastatic progression. These rare, self-renewing cells within tumors demonstrate remarkable plasticity and heterogeneity, driving the need for sophisticated molecular profiling techniques to accurately identify and study them [46]. Within the context of oncogenic potential assessment across stem cell types, researchers must select appropriate methodological approaches based on their specific experimental requirements, as each technology offers distinct advantages and limitations for stemness marker analysis.
Molecular profiling of stemness extends beyond mere identification to encompass the functional hierarchy within CSC populations. Evidence suggests that analogous to normal stem cell systems, CSCs exist in distinct functional states including Quiescent CSCs, Progenitor CSCs, and Progenitor-like CSCs, each characterized by unique marker expression profiles [46]. This hierarchical organization necessitates profiling techniques capable of resolving these subtle but biologically significant differences. The selection of an appropriate profiling method—whether PCR, flow cytometry, or reporter systems—depends fundamentally on the research objectives, whether for biomarker discovery, functional analysis, or high-throughput drug screening.
Technical Comparison of Stemness Profiling Methodologies
Performance Characteristics and Applications
Table 1: Comparative Analysis of Stemness Profiling Techniques
| Parameter | PCR-Based Methods | Flow Cytometry | Reporter Systems |
|---|---|---|---|
| Sensitivity | High (detects low abundance transcripts) | Moderate (limited by antibody affinity) | Variable (depends on promoter strength and detection method) |
| Throughput | High (96-384 well formats) | Moderate (single cell but limited by instrument speed) | High (compatible with live cell imaging and HTS) |
| Quantitative Capability | Excellent (absolute quantification possible) | Good (relative quantification based on fluorescence intensity) | Good (dynamic range varies by system) |
| Multiplexing Capacity | Moderate (4-8 targets with probe-based systems) | High (10-40 parameters with modern cytometers) | Limited (typically 1-3 reporters simultaneously) |
| Spatial Information | No (requires tissue homogenization) | No (single cell suspension required) | Yes (for imaging-compatible reporters) |
| Temporal Resolution | Endpoint measurement only | Endpoint measurement typically | Excellent (real-time monitoring possible) |
| Primary Application | Stemness gene expression profiling | Surface marker analysis and cell sorting | Functional assessment of promoter activity and cell tracking |
| Key Stemness Targets | OCT4, SOX2, NANOG, KLF4, c-MYC [47] [48] | CD44, CD133, CD90, EpCAM, ALDH activity [49] [46] | OCT4, SOX2, NANOG, ALDH1A1 promoters [48] [46] |
Workflow and Logical Relationships
Stemness Profiling Technology Selection Pathway
Polymerase Chain Reaction (PCR) Methods
Experimental Approach and Protocol
Reverse transcription quantitative PCR (RT-qPCR) represents the gold standard for sensitive, quantitative assessment of stemness-associated gene expression. The typical workflow involves RNA extraction from cell cultures or tissue samples, cDNA synthesis using reverse transcriptase, followed by amplification and quantification of target transcripts using sequence-specific primers and fluorescent probes [47]. Normalization to housekeeping genes is essential for accurate quantification, with data analysis typically performed using the comparative ΔΔCt method.
In a comprehensive study investigating CSC enrichment in colorectal cancer, researchers employed RT-qPCR to assess the upregulation of core stemness genes including KLF4, SOX2, OCT4, and c-MYC in 5-fluorouracil-resistant cells compared to parental cells [47]. The experimental protocol involved:
- Total RNA isolation using TRIzol reagent
- cDNA synthesis with a commercial kit
- Real-time PCR amplification using a Rotor-Gene Q LightCycler system
- Data analysis with normalization to reference genes
This approach demonstrated that drug-resistant cells exhibited significantly elevated expression of pluripotency factors, confirming the successful enrichment of CSC populations through chemotherapy exposure [47].
Research Reagent Solutions
Table 2: Essential Reagents for PCR-Based Stemness Analysis
| Reagent Category | Specific Examples | Function in Experiment |
|---|---|---|
| RNA Isolation | TRIzol reagent [47] | Maintains RNA integrity during extraction from cells/tissues |
| Reverse Transcription | cDNA synthesis kit [47] | Converts RNA to stable cDNA for amplification |
| Amplification System | SYBR Green or TaqMan probes [47] | Enables quantitative detection of amplified products |
| qPCR Instrument | Rotor-Gene Q LightCycler [47] | Provides precise thermal cycling and fluorescence detection |
| Stemness Primer Panels | OCT4, SOX2, NANOG, KLF4 primers [47] [48] | Target-specific amplification of key pluripotency factors |
Flow Cytometry Approaches
Experimental Design and Implementation
Flow cytometry enables multiparametric analysis of CSC populations at single-cell resolution based on surface marker expression and functional characteristics. The standard protocol involves creating a single-cell suspension, incubating with fluorochrome-conjugated antibodies against specific CSC markers, and analysis using a flow cytometer capable of detecting multiple fluorescence parameters simultaneously [50] [49]. For intracellular markers like transcription factors, cell permeabilization is required prior to antibody staining.
In colorectal cancer research, flow cytometric analysis confirmed elevated expression of CD44 and CD133 in 5-FU-resistant cells, validating their CSC-enriched status [47]. The experimental methodology included:
- Cell preparation using trypsin/EDTA treatment to create single-cell suspensions
- Antibody staining with fluorescently-labeled anti-CD44, anti-CD133, and anti-CD66 antibodies
- Data acquisition on a flow cytometer with appropriate laser and filter configurations
- Analysis and gating to identify and quantify positive populations
Advanced applications incorporate functional assays such as ALDH enzymatic activity measurement using fluorescent substrates like BODIPY-aminoacetaldehyde, which is converted to a fluorescent product by ALDH enzymes, identifying cells with high ALDH activity as potential CSCs [48] [49].
Research Reagent Solutions
Table 3: Essential Reagents for Flow Cytometric Stemness Analysis
| Reagent Category | Specific Examples | Function in Experiment |
|---|---|---|
| Surface Antibodies | Anti-CD44, CD133, CD90 [47] [49] | Detection of established CSC surface markers |
| Viability Stains | Propidium iodide [49] [51] | Exclusion of dead cells from analysis |
| ALDH Detection | ALDEFLUOR assay [48] [49] | Functional identification of stem cells via enzyme activity |
| Intracellular Staining Kits | Transcription factor staining buffers | Permeabilization for nuclear targets like OCT4, SOX2 |
| Compensation Beads | Anti-mouse/rat Ig κ compensation beads | Accurate color compensation for multicolor panels |
Reporter Gene Systems
Reporter gene systems constitute sophisticated molecular tools that leverage stemness-associated promoters to drive expression of easily detectable reporter proteins, enabling real-time monitoring and isolation of CSC populations. These systems typically employ fluorescent proteins (GFP, RFP) or enzymatic reporters (luciferase) under the control of regulatory elements from genes such as OCT4, SOX2, NANOG, or ALDH1A1 [48]. The fundamental advantage of this approach lies in its ability to dynamically track CSC populations without requiring cell fixation or destruction.
The design considerations for reporter systems include:
- Selection of appropriate regulatory elements - promoters versus enhancers
- Choice of reporter protein based on detection requirements
- Integration method - transient transfection versus stable integration
- Validation with established stemness markers and functional assays
As highlighted in recent reviews, promoter-based reporters for ALDH1A1 and pluripotency factors (OCT4, SOX2) demonstrate particular utility for marking heterogeneous CSC subpopulations, potentially enabling the discrimination between Quiescent CSCs, Progenitor CSCs, and Progenitor-like cells [46]. However, it is noteworthy that reporters for CD44 and CD133 are not generally recommended, as the functional activity of these markers is primarily regulated at the post-translational level rather than transcriptional level [46].
Research Reagent Solutions
Table 4: Essential Reagents for Reporter-Based Stemness Analysis
| Reagent Category | Specific Examples | Function in Experiment |
|---|---|---|
| Reporter Constructs | OCT4-GFP, SOX2-luciferase [48] [46] | Visualizing promoter activity of pluripotency factors |
| Transfection Reagents | Lipofectamine, electroporation kits | Introducing reporter constructs into target cells |
| Selection Antibiotics | Puromycin, G418 [48] | Selecting stably transfected cell populations |
| Live Cell Imaging Media | Phenol-red free media with HEPES | Maintaining cell viability during time-course imaging |
| Substrate Compounds | D-luciferin for luciferase systems | Generating detectable signals from enzymatic reporters |
Experimental Data and Comparative Performance
Quantitative Assessment from Published Studies
Table 5: Experimental Data Comparison Across Profiling Methodologies
| Experimental Context | PCR Findings | Flow Cytometry Findings | Reporter System Findings |
|---|---|---|---|
| Colorectal Cancer CSC Enrichment [47] | Upregulation of stemness genes: OCT4 (4.2-fold), SOX2 (3.8-fold), c-MYC (5.1-fold) in 5-FU resistant vs. parental cells | Increased CSC surface markers: CD44+ (28.7% vs 9.3%), CD133+ (35.2% vs 11.6%) in resistant vs parental cells | Not reported in this study |
| Gastric Cancer Stemness [52] [53] | Stemness signatures correlated with poor prognosis; NPC2 expression promoted stemness | CD51 identified as stemness regulator via flow cytometry; correlated with Notch activation | Not specifically reported |
| HCC Prognostic Markers [54] | Multiple stemness markers associated with outcomes | CK19/EpCAM co-expression: strongest predictor of OS (SUCRA: 78.65%); CD56 for poor differentiation | Not specifically reported |
| Breast Cancer CSC Hierarchy [46] | Used to validate subpopulation identities | CD44+/CD24- phenotype enriched in tumor-initiating cells | Dual reporters (pluripotency + ALDH1A1) recommended for heterogeneous CSCs |
Integrated Workflow for Comprehensive Stemness Assessment
Comprehensive Stemness Profiling Workflow
The comparative analysis of PCR, flow cytometry, and reporter systems for stemness marker profiling reveals a complementary relationship between these technologies rather than a competitive one. PCR-based methods provide superior sensitivity for quantifying transcriptional regulation of pluripotency factors, while flow cytometry enables isolation of viable CSC populations for functional studies based on surface markers and enzyme activity. Reporter systems offer unique capabilities for dynamic monitoring of stemness states and high-throughput screening applications.
For researchers designing studies on oncogenic potential assessment, the integration of multiple profiling approaches typically yields the most comprehensive understanding of CSC biology. An effective strategy might employ flow cytometry for initial population identification, followed by PCR-based validation of stemness signatures, and ultimately reporter system implementation for longitudinal studies and drug screening. This integrated approach leverages the respective strengths of each technology while mitigating their individual limitations, providing a robust framework for advancing stem cell research in oncological contexts.
The continuing evolution of these technologies—particularly the development of more sophisticated multiparameter flow cytometry and advanced reporter systems with in vivo imaging capabilities—promises to further enhance our ability to resolve the complex hierarchy and plasticity of cancer stem cells across different tumor types and experimental models.
The field of drug discovery has been transformed by the advent of human induced pluripotent stem cells (iPSCs), which provide a revolutionary platform for modeling human diseases and assessing compound toxicity. First discovered in 2006 by Shinya Yamanaka's lab, iPSCs are generated by reprogramming adult somatic cells into a pluripotent state through the introduction of specific transcription factors [55]. This breakthrough enabled researchers to create patient-specific stem cells capable of differentiating into virtually any cell type, bypassing both the ethical concerns of embryonic stem cells and the species limitations of animal models [56]. The FDA Modernization Act 2.0, implemented in December 2022, has further accelerated the adoption of iPSC technologies by advocating for alternative, non-animal testing methods including cell-based assays and organ-on-a-chip systems [57].
In the context of high-content screening (HCS), iPSC-derived cells offer unprecedented opportunities for detailed morphological and functional analysis of compound effects. High-content screening combines high-throughput automation with subcellular resolution microscopy, enabling quantitative observations of comprehensive phenotypes at the single-cell level [58]. This approach is particularly valuable for assessing complex neurodegenerative and cardiovascular diseases, where subtle changes in cellular morphology, protein aggregation, and organelle function serve as critical markers of disease progression and therapeutic efficacy [59] [58]. The integration of iPSC technology with advanced screening platforms addresses a fundamental challenge in drug development: the persistently high attrition rates where fewer than 10% of candidates entering clinical trials ultimately reach patients, with central nervous system programs failing up to 90% of the time [60].
Key Advantages of iPSC-Based Screening Platforms
Physiological Relevance and Human Disease Modeling
iPSC technology provides human-derived cellular models that closely replicate the genomic background and pathological mechanisms of human diseases. Unlike immortalized cell lines or animal primary cells, iPSC-derived models maintain patient-specific genetic variations, enabling more accurate recapitulation of disease phenotypes [60]. This human relevance is crucial for improving translational predictivity, as traditional models often fail to reliably forecast human outcomes due to species-specific differences in physiology, drug metabolism, and disease mechanisms [61] [57]. Research demonstrates that iPSC-derived cardiomyocytes (iPSC-CMs) accurately replicate cardiac pharmacological responses, providing superior models for predicting cardiotoxicity compared to animal models or conventional cell lines [57].
The patient-specific nature of iPSCs enables creation of comprehensive disease models for conditions including Alzheimer's disease, amyotrophic lateral sclerosis (ALS), Parkinson's disease, and various cardiac disorders [59] [56]. These models facilitate the study of disease mechanisms and compound effects in genetically relevant human cells, bridging the translational gap between preclinical research and clinical outcomes. For neurodegenerative diseases specifically, iPSC-derived neuronal cells allow researchers to investigate complex processes including neurite outgrowth, synaptic density, mitochondrial function, and protein aggregation in live human neurons [58].
Technical Advancements in High-Content Screening
Modern high-content screening platforms leverage cutting-edge microscopy and image analysis to extract rich quantitative data from iPSC-derived cultures. These systems typically employ high-throughput microplate imagers suitable for 24/96/384-well plates, with some advanced platforms utilizing confocal microscopes with spinning disk designs for increased resolution [58]. Super-resolution techniques such as structured illumination microscopy (SIM) and stimulated emission exhaustion microscopy (STED) enable investigation of subcellular structures beyond the diffraction limit, allowing researchers to characterize spatial organization of proteins in synapses or axon cytoskeleton architecture at nanometer scales [58].
The integration of artificial intelligence and machine learning with HCS has dramatically enhanced data analysis capabilities. AI algorithms excel at image recognition and can identify subtle disease-specific phenotypes in iPSC-derived cells, even from label-free microscopic images [58]. Machine learning technology enables computer systems to learn and predict responses from unknown datasets according to pre-trained programs, facilitating rapid analysis of large HCS datasets and detection of nuanced morphological patterns that might escape conventional analysis [58]. This AI-HCS combination is particularly valuable for drug screening applications, where it improves prediction of drug efficacy and toxicity while reducing analytical time and errors [62].
Table 1: Key Advantages of iPSC-Based High-Content Screening Platforms
| Feature | Advantage | Application in Drug Discovery |
|---|---|---|
| Human Relevance | Recapitulates human physiology and disease mechanisms more accurately than animal models | Improved prediction of drug efficacy and toxicity in human populations |
| Patient Specificity | Enables study of patient-specific disease variants and personalized drug responses | Development of personalized medicine approaches and identification of responder populations |
| High-Content Data | Provides multiparametric readouts at single-cell resolution | Deep phenotypic profiling of compound effects beyond simple viability assays |
| Genetic Engineering | Compatible with CRISPR/Cas9 gene editing for disease modeling and target validation | Creation of isogenic controls and precise investigation of gene function |
| Scalability | Suitable for high-throughput screening campaigns in 384-well formats | Screening of thousands of compounds in physiologically relevant human systems |
Comparative Analysis of iPSC Screening Platforms
Commercial iPSC Platform Providers
The growing adoption of iPSC technologies has spawned a competitive landscape of specialized companies providing iPSC-derived cells, differentiation protocols, and screening services. Ncardia has established itself as a leader in cardiac and neuronal applications, offering platforms such as the Ncyte Heart in a Box—3D cardiac microtissues designed for 384-well plate formats that enable screening of approximately 1,500 compounds per day [63]. Their solutions utilize 10-billion-cell batches of Ncyte vCardiomyocytes to enhance workflow efficiency and minimize revalidation efforts [63]. Similarly, FUJIFILM CDI (Cellular Dynamics International) manufactures multiple iPSC-derived cell types including iCell Cardiomyocytes, iCell Neurons, and iCell Hepatocytes, which are matched to function with common readout technologies and adapted to screening platforms [61].
bit.bio has developed innovative technology with their opti-ox precision cell programming platform, which enables deterministic conversion of iPSCs into defined human cell types within days at industrial scale [60]. Their ioCells product line provides highly consistent, cryopreserved iPSC-derived cells with exceptional purity, addressing the batch-to-batch variability issues that plague conventional differentiation protocols [60]. Other significant players include Evotec, with one of the largest and most advanced iPSC platforms globally, and Takara Bio, which offers standardized differentiation protocols for industrial-scale production of iPSC-derived hepatocytes that mimic embryonic development [61].
Table 2: Comparison of Leading Commercial iPSC Platform Providers
| Company | Core Technology/Specialization | Key Cell Types Available | Throughput Capabilities |
|---|---|---|---|
| Ncardia | 3D cardiac microtissues, high-throughput screening | Cardiomyocytes, neuronal cells | 1,500 compounds/day in 384-well format [63] |
| bit.bio | opti-ox precision cell programming | ioGlutamatergic Neurons, ioMicroglia, ioHepatocytes | Billions of consistent cells per manufacturing run [60] |
| FUJIFILM CDI | Large-scale manufacturing of iPSC-derived cells | Cardiomyocytes, neurons, hepatocytes, astrocytes | Multiple cell types adapted to screening platforms [61] |
| Evotec | Industrialized iPSC screening platform | Various specialized cell types | One of largest global iPSC platforms [61] |
| Takara Bio | Standardized differentiation protocols | Hepatocytes, cardiomyocytes | "Universal" protocol across multiple iPSC lines [61] |
Technical Specifications and Performance Metrics
When evaluating iPSC screening platforms, several technical parameters critically influence their performance in drug discovery applications. Assay reproducibility remains a fundamental challenge, with conventional directed differentiation protocols often producing variable cell populations that introduce noise into screening data [60]. Next-generation approaches like bit.bio's opti-ox technology address this by ensuring deterministic cell fate programming, resulting in less than 1% differential gene expression between lots [60]. Similarly, Ncardia's use of 10-billion-cell batches provides substantial consistency for extended screening campaigns [63].
The transition from 2D to 3D culture systems represents another significant advancement in platform capabilities. Three-dimensional microtissues better recapitulate native tissue architecture and function, providing more physiologically relevant models for compound testing [63]. For cardiac safety screening, 3D iPSC-derived cardiac microtissues demonstrate improved prediction of clinical cardiotoxicity compared to conventional 2D cultures, while simultaneously enabling higher throughput screening compatible with 384-well formats [63].
Platform versatility across applications is another key differentiator. Modern iPSC platforms support diverse readouts including high-content imaging, multi-electrode array (MEA) electrophysiology, calcium flux assays, and impedance-based monitoring [60]. This flexibility allows researchers to implement orthogonal assay approaches throughout the drug discovery workflow, from initial target identification and validation to lead optimization and safety profiling [60].
Experimental Design and Methodologies
High-Content Screening Workflow for iPSC-Based Assays
A robust high-content screening workflow for iPSC-based drug discovery integrates multiple specialized steps from cell culture to data analysis. The process typically begins with thawing and plating of cryopreserved iPSC-derived cells into microplates optimized for imaging, followed by a recovery period to allow cell attachment and functional maturation [60]. For neuronal applications, this may require 7-21 days to establish mature neurite networks and synaptic connections, while cardiomyocytes typically exhibit spontaneous beating within 7-14 days post-thaw [61] [60].
Compound treatment is performed using automated liquid handling systems to ensure precision and reproducibility across large compound libraries. Treatment duration varies based on the biological endpoint being measured—acute functional responses may require only minutes to hours, while chronic toxicity or disease-modifying effects might need days to weeks of compound exposure [58] [60]. For longitudinal studies, specialized live-cell imaging systems equipped with environmental chambers maintain optimal culture conditions throughout extended time courses [58].
Endpoint analysis in HCS typically involves cell fixation and staining with fluorescent markers targeting specific cellular structures or functions, followed by automated imaging across multiple wells and fields. Common staining panels for neurodegenerative disease models include markers for neuronal nuclei (NeuN), synaptic vesicles (SV2), microtubule-associated protein (MAP2) for neurites, and cleaved caspase-3 for apoptosis assessment [58]. For cardiac applications, stains for sarcomeric actinin, nuclei, and intracellular calcium are frequently employed to evaluate structural and functional endpoints [63] [57].
Diagram 1: High-content screening workflow for iPSC-based assays
Key Methodologies for Specific Applications
Neurite Outgrowth and Synaptic Density Analysis: For neurodegenerative disease research and neurotoxicity assessment, quantitative analysis of neurite outgrowth represents a critical HCS endpoint. The methodology involves immunostaining for neuronal-specific markers (βIII-tubulin, MAP2) and synaptic proteins (synaptophysin, PSD-95), followed by automated imaging and analysis using specialized software algorithms [58]. Open-source tools like CellProfiler and ImageJ with NeuriteTracer plugin enable automated tracing and quantification of neurite length, branching complexity, and synapse number [58]. These analyses provide insights into compound effects on neuronal network formation and maintenance, with applications in both neurodevelopmental toxicity testing and neurodegenerative drug discovery.
Cardiotoxicity Screening: iPSC-derived cardiomyocytes have become a standard tool for early cardiotoxicity assessment, particularly for pro-arrhythmic risk prediction. The Comprehensive in vitro Pro-arrhythmia Assay (CiPA) initiative has championed the use of iPSC-CMs alongside in silico modeling to improve prediction of clinical Torsades de Pointes risk [57]. Methodologies include high-speed calcium imaging to assess calcium handling, multi-electrode arrays (MEA) for field potential duration measurements, and impedance-based systems to monitor beating dynamics [57]. For high-content imaging, cardiomyocytes are stained with sarcomeric α-actinin to evaluate sarcomere organization, alongside live-cell dyes to monitor mitochondrial membrane potential and intracellular calcium transients [63] [57].
Hepatotoxicity Assessment: Drug-induced liver injury (DILI) remains a leading cause of drug attrition and post-market withdrawals, driving development of improved hepatotoxicity screening platforms. iPSC-derived hepatocytes offer a human-relevant system for DILI prediction, with methodologies including albumin and urea production assays, cytochrome P450 activity measurements, and high-content imaging of hepatic morphology [61] [60]. Multiparametric analysis typically includes assessment of cell viability, mitochondrial function, reactive oxygen species production, and intracellular lipid accumulation, providing a comprehensive profile of compound effects on hepatocyte health and function [60].
Research Reagent Solutions for iPSC-Based Screening
Successful implementation of iPSC-based high-content screening requires carefully selected reagents and tools optimized for stem cell applications. The following table details essential research reagent solutions for establishing robust screening platforms.
Table 3: Essential Research Reagent Solutions for iPSC-Based High-Content Screening
| Reagent Category | Specific Examples | Function & Application | Technical Considerations |
|---|---|---|---|
| iPSC-Derived Cells | iCell Cardiomyocytes (FUJIFILM CDI), ioGlutamatergic Neurons (bit.bio), Ncyte vCardiomyocytes (Ncardia) | Provide physiologically relevant human cells for screening; available as cryopreserved, ready-to-use formats | Batch-to-batch consistency, functional validation data, compatibility with screening formats [61] [63] [60] |
| Cell Culture Media | Specialty media optimized for specific cell types (neuronal, cardiac, hepatic) | Support maintenance and function of differentiated cells during screening assays | Formulation consistency, support of mature phenotype, compatibility with imaging applications |
| Cell Staining Reagents | Live-cell dyes (Calcein-AM, TMRM, Fluo-4), fixation-compatible antibodies | Enable multiplexed detection of cellular structures and functions | Photostability, compatibility with other fluorophores, minimal cellular toxicity |
| Analysis Software | CellProfiler, ImageJ with specialized plugins, Harmony (PerkinElmer) | Automated image analysis and quantification of high-content parameters | Algorithm accuracy, throughput capacity, customization options [58] |
| Microplates & Imaging Supplies | 384-well black-walled imaging plates, optical-quality lids | Provide optimal substrate for cell growth and high-quality image acquisition | Surface coating compatibility, autofluorescence levels, evaporation control |
Applications in Disease Modeling and Toxicity Assessment
Neurodegenerative Disease Modeling
iPSC-based screening platforms have demonstrated particular utility for modeling complex neurodegenerative disorders including Alzheimer's disease, ALS, and Parkinson's disease. Patient-derived iPSCs enable researchers to recapitulate disease-specific phenotypes in vitro, including protein aggregation, neurite retraction, synaptic dysfunction, and mitochondrial impairment [59] [58]. High-content imaging allows quantitative tracking of these pathological features across multiple parameters simultaneously, providing comprehensive profiling of disease progression and compound effects.
Clinical trials based on iPSC research have already been initiated for several neurodegenerative conditions. Examples include bosutinib, ropinirole and ezogabine for ALS, WVE-004 and BIIB078 for ALS with frontotemporal dementia (ALS/FTD), and bromocriptine for familial Alzheimer's disease [59]. These clinical translations highlight the growing impact of iPSC technology on therapeutic development for neurological disorders. Furthermore, the integration of iPSC-derived co-culture systems containing multiple CNS cell types (neurons, astrocytes, microglia) enables more sophisticated modeling of neuroinflammatory components in neurodegenerative diseases [60].
Cardiotoxicity and Hepatotoxicity Screening
Cardiotoxicity assessment represents one of the most established applications of iPSC technology in drug safety assessment. iPSC-derived cardiomyocytes provide a human-relevant system for detecting compound effects on cardiac repolarization, contractility, and structural integrity [57]. The CiPA initiative has extensively characterized reference compounds across multiple iPSC-CM platforms and assay technologies, demonstrating improved prediction of clinical pro-arrhythmic risk compared to traditional hERG channel assays alone [57]. Advanced 3D cardiac microtissue models further enhance physiological relevance while maintaining compatibility with high-throughput screening formats [63].
For hepatotoxicity assessment, iPSC-derived hepatocytes offer significant advantages over primary human hepatocytes, which suffer from limited availability, donor-to-donor variability, and rapid dedifferentiation in culture [61] [60]. Although functional maturity of iPSC-derived hepatocytes remains an area of active development, recent advances in differentiation protocols have yielded cells with robust drug metabolism capabilities and improved stability in culture [60]. These models enable detection of diverse hepatotoxicity mechanisms including steatosis, cholestasis, and mitochondrial toxicity through multiparametric high-content assays.
Diagram 2: iPSC applications in toxicity assessment across tissue types
Current Challenges and Future Directions
Technical Limitations and Solutions
Despite significant advances, iPSC-based screening platforms face several persistent challenges. Batch-to-batch variability remains a concern with conventional differentiation protocols, potentially introducing noise and reducing screening robustness [60]. Next-generation programming approaches like bit.bio's opti-ox technology address this through deterministic cell fate control, generating highly consistent cell populations with less than 1% differential gene expression between batches [60]. Similarly, Ncardia's production of 10-billion-cell batches provides substantial material for extended screening campaigns with minimal revalidation requirements [63].
Functional maturation of iPSC-derived cells represents another significant challenge, particularly for cell types that require extended development in vivo. iPSC-derived cardiomyocytes often exhibit fetal-like properties including spontaneous contraction and metabolic immaturity, while iPSC-derived neurons may lack the complete synaptic complexity of mature native neurons [57]. Advanced culture systems including 3D organoids, extended differentiation protocols, and electrical or mechanical stimulation strategies are being developed to enhance functional maturation [63] [57].
Scalability and throughput limitations have traditionally constrained implementation of complex iPSC-based models in high-throughput screening. However, recent technological advances are addressing these barriers. For example, Ncardia's Ncyte Heart in a Box platform enables screening of 1,500 compounds per day in 384-well format using 3D cardiac microtissues [63]. Similarly, bit.bio's manufacturing approach generates billions of consistently programmed cells in single production runs, supporting large-scale phenotypic screening campaigns [60].
Emerging Technologies and Future Outlook
The future of iPSC-based screening platforms will be shaped by several emerging technologies and trends. Artificial intelligence and machine learning are being increasingly integrated throughout the drug discovery workflow, from predicting differentiation outcomes to analyzing complex high-content imaging data [58] [62] [57]. AI-guided image analysis can identify subtle disease-relevant phenotypes that might escape conventional analysis, while machine learning algorithms improve prediction of compound efficacy and toxicity based on multiparametric screening data [62].
Multi-cell type co-culture systems and organoid models represent another frontier for iPSC technology, enabling more physiologically relevant modeling of tissue-level functions and cell-cell interactions [55]. These complex models better recapitulate native tissue architecture and function, though their implementation in high-throughput screening formats remains technically challenging. Ongoing developments in 3D microtissue production and analysis are gradually overcoming these barriers [63].
The regulatory landscape continues to evolve in support of human cell-based assays, with the FDA Modernization Act 2.0 accelerating the transition from animal models to human-relevant systems [57]. This regulatory shift, combined with continuing technological advances, suggests that iPSC-based screening platforms will play an increasingly central role in drug discovery and toxicity assessment throughout the pharmaceutical industry. As these platforms mature, they promise to improve the efficiency and success rates of therapeutic development while reducing reliance on animal models.
The assessment of oncogenic potential across different stem cell types is a critical challenge in regenerative medicine and cancer biology. Traditional bulk analysis methods, which average signals across entire cell populations, often mask the cellular heterogeneity that is fundamental to understanding cancer initiation and progression. The convergence of microfluidics, single-cell genomics, and artificial intelligence (AI) has created a transformative paradigm for identifying and quantifying cancer risk with unprecedented resolution. This technological triad enables researchers to deconvolute complex biological systems by investigating individual cells, revealing rare but biologically significant subpopulations that drive disease progression and therapeutic resistance [64].
Within seemingly homogeneous stem cell populations, cellular heterogeneity arises from genetic, epigenetic, and environmental variations. Even within clonal populations, stochastic gene expression and dynamic cellular states contribute to variability in function and fate [64]. The integration of microfluidic single-cell isolation with high-throughput genomic profiling and AI-driven computational analysis provides a powerful framework for identifying stem cell subpopulations with elevated oncogenic potential, mapping their molecular signatures, and predicting their behavioral trajectories. This approach is particularly valuable for evaluating the safety of stem cell-based therapies, where understanding and quantifying cancer risk is paramount for clinical translation [65].
Technology Platform Comparisons
The selection of appropriate technological platforms is crucial for designing robust experiments aimed at assessing oncogenic potential. The table below provides a systematic comparison of major high-throughput single-cell RNA sequencing platforms, highlighting their performance characteristics based on experimental data from peer-reviewed studies.
Table 1: Performance Comparison of High-Throughput Single-Cell RNA Sequencing Platforms
| Platform | Cell Throughput | Sensitivity (Genes/Cell) | Doublet Rate | Key Strengths | Best Applications in Stem Cell Research |
|---|---|---|---|---|---|
| 10x Genomics Chromium X | High (10,000-100,000 cells) | ~1,000-5,000 genes/cell [66] | 0.4-11% (cell loading-dependent) [67] | High throughput, extensive gene panels (up to 5,000-plex) [68] | Large-scale stem cell heterogeneity studies, rare population identification |
| MobiNova-100 (MobiDrop) | High (Comparable to Chromium X) | Performance close to Chromium X [66] | Similar to other droplet-based systems | Excellent differential gene expression significance [66] | Oncogenic driver gene identification, stem cell differentiation studies |
| SeekOne (SeekGene) | Variable | Good sensitivity | Standard for droplet platforms | Customizable workflows | General stem cell transcriptome mapping |
| C4 (BGI) | Variable | Good sensitivity | Standard for droplet platforms | Cost-effective solutions | Large-scale screening applications |
Platform selection significantly impacts the ability to resolve subtle differences in stemness signatures and oncogenic trajectories. Each system employs droplet-based microfluidics to partition individual cells into oil emulsions, enabling massively parallel barcoding and sequencing [67]. However, they differ in their sensitivity to detect rare transcripts, ability to minimize multiplets (droplets containing more than one cell), and overall reproducibility—all critical factors when evaluating stem cell populations for potentially oncogenic subpopulations that may represent less than 1% of the total population [64].
Experimental Frameworks for Oncogenic Risk Assessment
Validating Single-Cell Measurements: Species Mixing and Doublet Detection
A critical consideration in single-cell genomics is ensuring that measurements truly represent individual cells rather than technical artifacts. The species-mixing experiment serves as the gold-standard validation approach, where human and mouse cells are mixed together in known ratios prior to processing [67]. After sequencing, bioinformatic analysis identifies "heterotypic doublets"—droplets containing cells from both species—which appear as hybrid cells expressing both human and mouse genes in what is known as a "barnyard plot" [67].
Table 2: Experimental Protocols for Key Single-Cell Validation Methods
| Method | Experimental Design | Key Procedures | Data Interpretation | Application in Oncogenic Risk Assessment |
|---|---|---|---|---|
| Species Mixing | Combine human and mouse cells at 50:50 ratio [67] | Process mixed cell population through single-cell platform | Identify heterotypic doublets via mixed-species expression profiles; calculate total doublet rate [67] | Ensures rare cancer stem cell populations are not technical artifacts |
| Cell Hashing/MULTI-seq | Label distinct cell populations with unique oligonucleotide barcodes (antibody-derived or lipid-conjugated) [67] | Pool barcoded populations before loading onto platform | Detect multiple barcodes per droplet to identify doublets; enables sample multiplexing [67] | Allows tracking of multiple stem cell lines or differentiation timepoints in one experiment |
| Ambient RNA Quantification | Analyze empty droplets containing only background solution [67] | Sequence all droplets, including those without cells | Profile background RNA signal using computational tools like SoupX or DecontX | Prevents misclassification of stem cells based on contaminating RNA signals |
For stem cell research, particularly when investigating rare cancer stem cell (CSC) populations, controlling doublet rates is essential since these rare cells can be misinterpreted as novel transitional states when in fact they represent technical artifacts [67]. Cell hashing approaches with barcoded antibodies (CITE-seq) or lipid-conjugated oligonucleotides (MULTI-seq) enable not only doublet identification but also sample multiplexing, allowing researchers to pool multiple stem cell lines or differentiation timepoints in a single sequencing run, thereby reducing batch effects and improving experimental consistency [69] [67].
Microfluidic Workflows for Single-Cell Analysis
Microfluidics provides the physical foundation for high-throughput single-cell analysis through precise manipulation of fluids at the microscale. The workflow begins with single-cell separation, followed by cell lysis, nucleic acid amplification, and library preparation—all potentially integrated within a microfluidic device [70].
Single-Cell Separation Methods:
- Hydrodynamic cell traps: Use channel geometry to physically capture individual cells [70]
- Pneumatic membrane valves: Employ controlled pressure to isolate cells in microchambers [70]
- Droplet-based isolation: Encapsulate single cells in water-in-oil emulsions, creating discrete reaction vessels [64] [70]
Droplet-based microfluidics has emerged as the dominant approach for high-throughput applications, enabling massive parallelization with throughputs of up to 44,000 single cells in a single run [64]. This method forms the basis for commercial platforms like 10x Genomics Chromium and facilitates applications such as Drop-seq and CITE-seq [64] [69]. The miniaturized volumes (picoliter to nanoliter) significantly reduce reagent costs while maintaining high sensitivity—a crucial advantage when working with precious stem cell samples [69].
Following cell isolation, effective lysis and nucleic acid amplification are critical steps. Microfluidic approaches minimize lysate dilution, enhancing detection sensitivity for low-abundance transcripts that may serve as early indicators of oncogenic transformation [70]. Whole-transcriptome amplification (WTA) and whole-genome amplification (WGA) methods have been optimized for microfluidic formats, providing sufficient material for downstream sequencing while preserving representation of original nucleic acid populations [70].
AI-Driven Stemness Quantification and Risk Prediction
Computational Frameworks for Stemness Assessment
Artificial intelligence, particularly machine learning (ML) and deep learning (DL), has revolutionized the interpretation of single-cell genomic data by identifying complex, non-linear patterns that escape conventional statistical methods. In stem cell research, AI approaches have been successfully applied to quantify stemness indices—numerical representations of the number and activity of stem cells within a population [71].
Multiple stemness indices have been developed:
- mRNAsi: Based on transcriptomic signatures of stemness [71]
- mDNAsi: Derived from DNA methylation patterns associated with stem cell states [71]
- DMPsi: Incorporating differential methylated regions [71]
- ENHsi: Utilizing enhancer element activity profiles [71]
These indices serve as crucial biomarkers for oncogenic potential assessment, as they capture the molecular resemblance between normal stem cells and cancer stem cells (CSCs)—the subpopulation responsible for tumor initiation, progression, and therapy resistance [71]. AI models can integrate these multi-omics measurements to generate comprehensive stemness profiles that predict the likelihood of malignant transformation.
Machine Learning for Clinical Outcome Prediction
Beyond stemness quantification, AI systems show remarkable promise in predicting clinical outcomes relevant to stem cell therapies. A recent study demonstrated the application of machine learning and explainable AI (XAI) techniques to predict survival rates in children undergoing hematopoietic stem cell transplants [72]. The custom stacked model achieved impressive performance metrics with 89% precision, 88% recall, 88% F1-score, and 92% AUC [72].
Explainable AI techniques including SHAP (Shapley Additive Values), LIME (Local Interpretable Model-agnostic Explanations), ELI5, and QLattice identified relapse, donor age, recipient age, and platelet recovery time as the most important predictive features [72]. This approach exemplifies how AI can transform complex molecular and clinical data into actionable insights for risk assessment—a framework that can be adapted for evaluating oncogenic potential in stem cell therapies.
The Scientist's Toolkit: Essential Research Solutions
Table 3: Key Research Reagent Solutions for Single-Cell Oncogenic Risk Assessment
| Product Category | Specific Examples | Function in Workflow | Application in Stem Cell Research |
|---|---|---|---|
| Droplet-Based scRNA-seq Kits | 10x Genomics Chromium Single Cell Gene Expression, Parse Biosciences Single-Cell Product Line [68] | Partitioning cells, barcoding transcripts, library preparation | High-throughput transcriptome profiling of stem cell populations |
| Multi-omics Profiling Kits | CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) [69] [67] | Simultaneous measurement of mRNA and surface proteins | Identifying cancer stem cell surface markers alongside gene expression |
| Cell Hashing Reagents | BioLegend TotalSeq Antibodies, MULTI-seq Lipids [67] | Sample multiplexing and doublet detection | Pooling multiple stem cell lines or timepoints in one experiment |
| Spatial Transcriptomics Kits | 10x Genomics Xenium Gene Panels, Slide-seq [69] | Spatial mapping of gene expression in tissue context | Localizing putative oncogenic stem cells in complex tissue architectures |
| Automated Cell Culture Systems | Robotic operation systems (e.g., Maholo) [64] | Standardized cell maintenance and differentiation | Reducing technical variability in stem cell culture prior to analysis |
The integration of microfluidics, single-cell genomics, and AI represents a paradigm shift in how researchers approach oncogenic risk assessment in stem cell populations. This technological convergence enables unprecedented resolution in detecting rare subpopulations, quantifying stemness signatures, and predicting malignant potential. As these technologies continue to mature and become more accessible through commercial solutions and cloud-based platforms [64] [73], they promise to accelerate the development of safer stem cell-based therapies while providing fundamental insights into the mechanisms of cancer initiation.
Visualizing Experimental Workflows and Signaling Pathways
Single-Cell RNA-Sequencing Workflow
Single-Cell RNA-Sequencing Workflow from Sample to Stemness Index
Stemness Signaling Pathway Regulation
Stemness Signaling Pathways in Normal and Oncogenic States
Navigating Challenges: Sensitivity Limits, Heterogeneity, and Standardization Hurdles
The therapeutic potential of stem cells in regenerative medicine is immense, yet their clinical application is tempered by the persistent risk of oncogenic transformation. The presence of undifferentiated pluripotent stem cells in cell therapy products can lead to teratoma formation or malignant tumor development, making the establishment of sensitive and specific detection thresholds a critical component of biosafety assessment [7]. For researchers and drug development professionals, navigating the landscape of detection technologies requires a clear understanding of performance trade-offs between sensitivity, specificity, and practical implementation. This guide provides an objective comparison of current methodologies, supported by experimental data, to inform selection of appropriate detection strategies for oncogenic potential assessment across diverse stem cell types.
Comparative Analysis of Detection Methodologies
The table below summarizes the performance characteristics of key technologies used for detecting oncogenic potential and residual undifferentiated stem cells:
Table 1: Performance Comparison of Detection Methodologies
| Methodology | Theoretical Sensitivity | Reported Sensitivity | Specificity | Sample Input | Throughput |
|---|---|---|---|---|---|
| RT-LAMP | 0.00002% hiPSC contamination | 0.00002% hiPSC contamination [74] | High (recognizes 6 target regions) | Microgram-order RNA per test [74] | High (one-pot reaction) |
| Deep Learning Morphological Analysis | N/A | AUC: 0.975, Accuracy: 0.922, Specificity: 0.942 [75] | Specificity: 0.942 [75] | Live-cell images | High (automated) |
| Flow Cytometry Purity Assessment | Dependent on antibody specificity and cell numbers | Not explicitly quantified | Dependent on antibody specificity [76] | 1×10^6 to 1×10^7 cells/mL [76] | Medium |
| Proteomic Stemness Index (PROTsi) | Protein-expression based | Correlates with aggressive tumor subtypes [14] | Identifies tumor-specific and shared targets [14] | Protein samples | Medium |
| T-cell Reference WES | Sensitivity: 0.91-1.00 [77] | 0.91-1.00 | Low false positive rate (average 1.5 false positives) [77] | DNA for whole exome sequencing | Low |
Detailed Experimental Protocols
RT-LAMP for Pluripotency Marker Detection
The Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) protocol offers exceptional sensitivity for detecting residual undifferentiated human pluripotent stem cells (hPSCs) in differentiated cell products [74].
Sample Preparation:
- Spike known numbers of undifferentiated hiPSCs (e.g., Ff-I01s04 line) into differentiated cell populations (hepatocytes, endothelial cells, neural progenitor cells)
- Pellet mixed cell populations by centrifugation
- Extract total RNA using commercial kits (e.g., PureLink RNA Mini Kit) with elution in distilled water
- Determine RNA concentration and quality via spectrophotometry (A260/A230 ratio) [74]
RT-LAMP Reaction Setup:
- Design LAMP primers to recognize 6 distinct regions of pluripotency marker genes (e.g., NANOG, POU5F1)
- Include loop primers to accelerate amplification kinetics
- Prepare reaction mixture containing thermostable Bst DNA polymerase with strand displacement activity
- Add thermostable reverse transcriptase for one-pot RT-LAMP
- Use microgram quantities of total RNA per test to enhance detection sensitivity
- Perform amplification at isothermal conditions (60-65°C) for 30-60 minutes [74]
Detection and Analysis:
- Monitor amplification in real-time using intercalating dyes or turbidity measurement
- Confirm specificity with gel electrophoresis or colorimetric indicators
- Calculate detection sensitivity based on standard curves from spiked samples
- Validate against multiple hPSC lines (e.g., Ff-I01, 1383D6, 1231A3) to ensure robustness [74]
Deep Learning for Morphological-Based Screening
This protocol uses convolutional neural networks (CNNs) to classify mesenchymal stem cell lines based on morphological features correlated with stemness markers [75].
Cell Culture and Imaging:
- Culture human neural crest-derived nasal turbinate stem cells (hNTSCs) in α-MEM with 10% FBS
- Maintain at 37°C in 5% CO₂ with medium changes every 2-3 days
- Harvest cells at passage 6 for consistency
- Seed 1×10^6 cells in 6-well culture plates for imaging
- Acquire live-cell microscopic images using automated microscope (e.g., Lionheart LX)
- Capture images at 40× and 100× magnification as 8-bit grayscale (904×1224 pixels) [75]
Image Preprocessing:
- Exclude overexposed images (mean value >230)
- Adjust remaining images to mean value of 130 for consistency
- Resize images to 226×306 pixels using interpolation in Python OpenCV
- Normalize pixel values to [0, 1] range
- Apply data augmentation techniques to expand training dataset [75]
Model Training and Validation:
- Implement transfer learning with pretrained CNN models (DenseNet121, VGG19, ResNet50V2)
- Replace final classification layers with task-specific layers
- Train models to classify MSC lines as high/low multilineage differentiating stress-enduring (MUSE) cells
- Validate classifications against immunofluorescence staining for SSEA-3 and CD-105 markers
- Use FACS analysis as ground truth for MUSE cell quantification
- Evaluate model performance using AUC, accuracy, F1 score, sensitivity, and specificity metrics [75]
Flow Cytometry for Purity Assessment
This protocol ensures accurate assessment of isolated cell population purity, critical for reliable lineage-specific chimerism analysis [76].
Cell Staining:
- After cell separation, aliquot 100μL of enriched cells (1×10^6 to 1×10^7 cells/mL) into FACS tubes
- Add fluorochrome-conjugated monoclonal antibodies against target surface markers (e.g., CD3 for T cells, CD19 for B cells)
- Include appropriate isotype control antibodies in separate tube
- Optional: Add viability stain (propidium iodide or 7AAD) to exclude dead cells
- Incubate at 2-8°C for 30 minutes in the dark
- Wash cells with PBS, resuspend in PBS or FACS sheath fluid
- Fix with 1% paraformaldehyde if not analyzing immediately [76]
Flow Cytometry Analysis:
- Create dot plot displaying FSC vs. SSC
- Gate around leukocytes to exclude RBC and debris
- Create second dot plot of FSC vs. viability stain
- Gate to exclude dead cells positive for viability marker
- Collect 10,000-50,000 events per sample
- Calculate purity as percentage of cells positive for relevant staining antibody in gated population
- Document purity assessment results following EFI and ASHI guidelines [76]
Visualization of Methodologies
Detection Workflow Comparison
Decision Framework for Methodology Selection
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Oncogenic Potential Assessment
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cell Isolation Kits | EasySep, RoboSep | Isolation of specific cell lineages for purity assessment | Follow manufacturer's PIS for purity protocols [76] |
| Pluripotency Markers | SSEA-3, CD105, NANOG, POU5F1 | Detection of undifferentiated stem cells | Marker expression varies across cell types [8] [75] |
| Antibody Panels | CD3, CD19, CD34, CD45, CD73, CD90 | Cell surface profiling and purity assessment | Use fluorochrome-conjugated antibodies for flow cytometry [76] |
| Nucleic Acid Amplification | RT-LAMP primers, Bst polymerase | Sensitive detection of pluripotency marker RNAs | Design primers for 6 target regions for specificity [74] |
| Cell Culture Media | α-MEM with FBS, StemFit AK02N | Maintenance of stem cell cultures | Use consistent media for reproducible results [75] |
| Imaging Reagents | Paraformaldehyde, DAPI, viability stains | Cell fixation, nuclear staining, and viability assessment | Include viability stains to gate out dead cells [75] [76] |
The establishment of clinically relevant detection thresholds requires careful consideration of the complementary strengths and limitations of available technologies. RT-LAMP provides exceptional sensitivity for minimal residual undifferentiated cell detection, while deep learning approaches offer high-throughput functional assessment based on morphological profiling. Flow cytometry remains the gold standard for purity assessment of specific cell populations, and emerging proteomic indices show promise for quantifying stemness characteristics associated with oncogenic potential. For researchers developing stem cell therapies, a layered approach combining multiple methodologies may provide the most comprehensive safety assessment, balancing the competing demands of sensitivity, specificity, and practical implementation in clinical manufacturing settings. As the field advances, continued refinement of these detection thresholds will be essential for ensuring patient safety while enabling the therapeutic potential of stem cell-based regenerative medicine.
Cancer stem cells (CSCs) represent a subpopulation of tumor cells with self-renewal capacity and the ability to drive tumor growth, metastasis, and relapse. These cells, often constituting less than 5% of the total cancer cell pool, are widely recognized as major contributors to therapeutic resistance [78] [8]. The traditional view of CSCs as static, marker-defined entities has been fundamentally challenged by recent technological advances. Emerging evidence from single-cell sequencing studies suggests that stemness might be a dynamic, context-dependent state rather than a fixed cellular phenotype [78] [3]. This paradigm shift necessitates increasingly sophisticated detection strategies to capture the full complexity of CSC biology, particularly given their critical role in treatment failure across epithelial malignancies including pancreatic ductal adenocarcinoma, triple-negative breast cancer, and colorectal cancer [78].
The detection and characterization of rare CSCs presents unique challenges for researchers. Cellular plasticity enables transitions between stem and non-stem states, while the absence of universal CSC markers complicates isolation and tracking efforts [8] [3]. Furthermore, CSCs often exist in a quiescent state, employing dormancy as a survival mechanism against conventional therapies that target rapidly dividing cells [79]. This review systematically compares current methodologies for rare CSC and undifferentiated cell detection, providing experimental protocols, quantitative performance data, and analytical frameworks to guide research in oncogenic potential assessment across stem cell types.
Comparative Analysis of CSC Detection Methodologies
Technologies for Identification and Isolation
Table 1: Comparison of Core CSC Detection and Analysis Technologies
| Method Category | Specific Technology | Key Measurable Parameters | Detection Sensitivity | Throughput | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|---|
| Marker-Based Isolation | Fluorescence-Activated Cell Sorting (FACS) | Surface marker expression (CD44, CD133, CD24) | ~0.1% of population [3] | Medium | High purity; viable cells for functional assays | Marker expression variability and instability |
| Functional Assays | Tumorsphere Formation | Self-renewal capacity; primary and secondary sphere formation efficiency | Varies by cancer type [8] | Low | Measures functional stemness without marker dependence | Microenvironment influences; semi-quantitative |
| Metabolic Profiling | ALDH Activity Detection (ALDEFLUOR) | ALDH enzyme activity | <1% of population [3] | Medium | Identifies metabolically distinct CSCs; works with viable cells | Tissue-specific variability in utility |
| Single-Cell Omics | scRNA-seq with RNA Velocity | Transcriptional heterogeneity; trajectory inference; stemness scores | Rare population resolution (<0.1%) [78] | High | Unbiased characterization; dynamic state prediction | High cost; computational complexity |
| Computational Inference | Stemness Scoring Algorithms (CytoTRACE, mRNAsi) | Differentiation state; gene expression entropy | Single-cell resolution [78] | High | Marker-independent; works with existing datasets | Reference-dependent; validation required |
Computational Stemness Assessment Tools
Table 2: Bioinformatics Tools for CSC Characterization and Stemness Quantification
| Tool Name | Algorithm Basis | Platform Availability | Input Requirements | Key Applications | Technical Considerations |
|---|---|---|---|---|---|
| CytoTRACE/CytoTRACE2 | Gene counts and expression diversity; deep learning | R, Python, web server [78] | scRNA-seq expression matrix | Predicts differentiation status; identifies progenitor-like cells | Sensitive to data quality and normalization |
| mRNAsi | Machine learning reference to pluripotent stem cells | R, web server [78] | Bulk or single-cell RNA-seq data | Quantifies oncogenic dedifferentiation; pan-cancer stemness index | Training set dependency; may miss context-specific states |
| StemID | Shannon entropy calculation | R [78] | scRNA-seq data | Lineage inference; stemness probability estimation | Requires sufficient cell numbers for robust entropy calculation |
| SCENT | Signaling entropy and connectome correlation | R [78] | scRNA-seq with or without network data | Cellular potency assessment; integrates transcriptional networks | Computational intensity for large networks |
| PROTsi | Protein expression patterns | Not specified [14] | Proteomic data | Quantifies stem-like features from proteomic data; links to clinical outcomes | Limited to available proteomic datasets |
Experimental Protocols for CSC Detection and Validation
Integrated Single-Cell RNA Sequencing Workflow
The standardized single-cell processing workflow enables high-resolution profiling of rare CSC subsets through reproducible bioinformatics pipelines [78]. The following protocol outlines the complete process from sample preparation to stemness assessment:
Sample Processing and Library Preparation:
- Begin with fresh or properly preserved tissue samples (tumor biopsies or liquid biopsies). Dissociate tissues into single-cell suspensions using enzymatic digestion optimized for the specific tissue type (e.g., collagenase/hyaluronidase mixtures for solid tumors).
- Remove dead cells and debris using density gradient centrifugation or magnetic bead-based separation. Assess cell viability (>90% recommended) using trypan blue or fluorescent viability dyes.
- For droplet-based scRNA-seq platforms (10X Genomics, Drop-seq), concentrate cells to optimal density (700-1,200 cells/μL). Prepare barcoded gel beads and partitioning oil according to manufacturer specifications.
- Generate single-cell transcriptome libraries following platform-specific protocols, typically involving reverse transcription, cDNA amplification, and library construction with sample indices.
Quality Control and Bioinformatics Processing:
- Sequence libraries to a minimum depth of 50,000 reads per cell on Illumina platforms. Perform initial quality control using FastQC to assess sequencing quality, adapter contamination, and other potential issues.
- Align reads to the appropriate reference genome (e.g., GRCh38) using splice-aware aligners like STAR. Generate gene expression matrices with unique molecular identifier (UMI) counting using Cell Ranger or similar pipelines.
- Filter low-quality cells using thresholds for mitochondrial gene percentage (<20%), minimum gene detection (>200 genes/cell), and doublet detection. Normalize data using SCTransform or similar methods to account for technical variability.
Stemness Assessment and CSC Identification:
- Perform dimensionality reduction using PCA followed by UMAP or t-SNE visualization. Cluster cells using graph-based methods (Louvain, Leiden) at multiple resolutions to identify distinct subpopulations.
- Calculate stemness scores using computational tools such as CytoTRACE2 (predicts differentiation status based on gene counts diversity) or integrate multiple algorithms through Cancer Stemness Online platform [78].
- Identify candidate CSC populations as clusters with high stemness scores and expression of known CSC markers (combination of canonical and context-dependent markers). Validate functional potential through downstream assays.
Functional Validation Using Tumorsphere Formation Assay
The tumorsphere formation assay assesses self-renewal capacity under non-adherent conditions, a hallmark functional property of CSCs [8] [3]:
Primary Sphere Formation:
- Prepare serum-free DMEM/F12 medium supplemented with B27 (1:50), epidermal growth factor (20 ng/mL), basic fibroblast growth factor (20 ng/mL), and heparin (4 μg/mL).
- Plate single-cell suspensions at clonal density (500-1,000 cells/cm²) in ultra-low attachment plates to prevent adhesion and encourage sphere formation.
- Culture for 7-14 days at 37°C with 5% CO₂, monitoring sphere formation every 2-3 days. Add fresh growth factors twice weekly without disturbing forming spheres.
- After 7-14 days, quantify spheres >50μm in diameter using inverted microscopy. Calculate sphere formation efficiency as (number of spheres formed / number of cells seeded) × 100%.
Secondary and Tertiary Sphere Formation:
- Collect primary spheres by gentle centrifugation (800 rpm for 5 minutes). Dissociate into single cells using enzymatic (trypsin/accutase) or mechanical means.
- Replate dissociated cells at the same clonal density in fresh sphere-forming medium. Culture for an additional 7-14 days.
- Quantify secondary sphere formation. True CSCs will demonstrate self-renewal capacity through serial passaging with maintained or increased sphere-forming efficiency.
Stemness Validation:
- For marker-based validation, harvest spheres for FACS analysis of established CSC markers (CD44+/CD24- for breast cancer, CD133 for glioblastoma, etc.) [3].
- Correlate sphere-forming efficiency with computational stemness predictions from scRNA-seq data to validate bioinformatic approaches.
- Compare gene expression profiles of sphere-derived cells versus adherent cultures using qRT-PCR for stemness genes (OCT4, NANOG, SOX2).
Signaling Pathways Governing CSC Stemness and Detection Applications
The molecular regulation of CSC maintenance involves complex signaling networks that provide both detection targets and therapeutic opportunities. The following diagram illustrates the core pathways and their crosstalk in CSC regulation:
Multiple interconnected signaling pathways regulate CSC maintenance and plasticity. The WNT/β-catenin, Hedgehog, and Notch pathways constitute core stemness regulation networks, while NF-κB, JAK/STAT, TGF-β, and PI3K/AKT pathways provide contextual signaling input [3] [80]. These pathways converge to regulate fundamental CSC properties including epithelial-mesenchymal transition (EMT), quiescence, and metabolic reprogramming. Pathway activity monitoring provides a functional readout of stemness states independent of surface markers, with specific applications in detection strategy design. For instance, TGF-β pathway activation promotes EMT, enhancing stem-like properties and invasive capacity through transcription factors such as ZEB1, SNAIL, and TWIST [80]. Similarly, PI3K/AKT signaling drives metabolic shifts toward glycolysis, supporting CSC survival in hypoxic tumor niches. The detection of pathway activity through phospho-flow cytometry, reporter assays, or transcriptional targets offers complementary approaches to identify CSCs based on functional signaling states rather than static marker expression.
Essential Research Reagent Solutions for CSC Detection
Table 3: Key Research Reagents for CSC Detection and Characterization Experiments
| Reagent Category | Specific Examples | Primary Applications | Technical Considerations | Commercial Sources |
|---|---|---|---|---|
| Surface Marker Antibodies | Anti-CD44, Anti-CD133, Anti-CD24, Anti-CD34, Anti-CD38 | FACS isolation; immunofluorescence validation | Lot-to-lot variability requires validation; species compatibility critical | BD Biosciences, BioLegend, Miltenyi Biotec |
| Intracellular Staining Kits | ALDEFLUOR Kit (ALDH activity) | Metabolic CSC identification; flow cytometric analysis | Requires specific inhibitor control; cell viability affects results | STEMCELL Technologies |
| Stemness Pathway Reporters | TOPFlash (WNT), Gli-luciferase (Hedgehog), CSL-luciferase (Notch) | Pathway activity quantification; functional CSC monitoring | Transfection efficiency variable; may require stable cell line generation | Addgene, commercial reporter constructs |
| 3D Culture Matrices | Ultra-low attachment plates; Matrigel; defined hydrogels | Tumorsphere assays; stemness maintenance in culture | Batch variability in natural matrices; defined matrices improve reproducibility | Corning, Thermo Fisher Scientific |
| Single-Cell Analysis Kits | 10X Genomics Chromium; SMART-seq kits | scRNA-seq library preparation; transcriptional stemness assessment | Cell viability critical; input cell concentration optimization needed | 10X Genomics, Takara Bio |
| Viability Dyes | Propidium iodide; 7-AAD; DAPI; viability dyes for FACS | Dead cell exclusion in sorting and analysis | Compatibility with other fluorochromes; DNA-binding dyes preclude downstream DNA analysis | Multiple suppliers |
The detection of rare cancer stem cells and undifferentiated cell populations requires multimodal approaches that address both technical and biological challenges. No single methodology sufficiently captures the dynamic nature of CSC states, necessitating integrated strategies that combine surface marker detection, functional assays, metabolic profiling, and computational assessment. The evolving paradigm of stemness as a transient, context-dependent property rather than a fixed identity underscores the importance of dynamic single-cell technologies and trajectory inference algorithms [78].
Future directions in CSC detection will likely focus on spatial transcriptomics to resolve niche-specific stemness programs, multi-omics integration to connect transcriptional and proteomic stemness signatures, and functional CRISPR screens to validate detection predictions [78] [14]. For researchers assessing oncogenic potential across stem cell types, the strategic combination of computational stemness scoring with functional validation provides the most robust framework for identifying and characterizing these critical drivers of tumor progression and therapeutic resistance. The continued refinement of CSC detection methodologies will enable more precise targeting of these elusive populations, ultimately contributing to improved outcomes in cancer therapy.
The biomedical research field faces a significant reproducibility crisis, with concerns that many published findings cannot be reliably replicated in independent laboratories [81]. In oncology and stem cell research, this problem is particularly acute, where technical variability in assays can lead to inconsistent results that hamper scientific progress and clinical translation [82] [81]. The inability to reproduce findings has had tangible consequences, including the halting of cancer clinical trials when key molecular signatures used for decision-making failed independent validation [82].
This challenge stems from multiple sources throughout the experimental workflow. With the development of novel high-throughput technologies, experiments have become increasingly complex, involving instruments sensitive to specific settings and generating massive datasets that require sophisticated processing [82]. Variability can be introduced at any stage—from biological sample preparation and reagent batches to data analysis procedures—making it difficult for independent researchers to reproduce published studies [82]. In cancer stem cell research, where accurately assessing oncogenic potential across different stem cell types is crucial, this lack of reproducibility directly impacts our understanding of tumor initiation, progression, and therapeutic resistance [83] [84].
Fundamental Concepts: Accuracy, Precision, and Their Implications
Understanding comparability and reproducibility (C&R) requires clarity on their foundational metrics: accuracy and precision. Accuracy indicates how close a measurement is to its true value, while precision indicates how close repeated measurements are to each other [82]. Deviation from accuracy (bias) often stems from systematic error sources that cannot be removed by simply repeating measurements, whereas precision (variability) can generally be improved by increasing the number of measurements [82].
In practice, there is often a trade-off between accuracy and precision [82]. For example, in microarray image analysis, foreground intensities are typically less variable but can exhibit higher bias compared with background-corrected intensity [82]. This relationship has crucial implications for oncogenic potential assessment, as reproducible data do not necessarily require unbiased measurements as long as they are "consistently inaccurate" [82]. However, consistently inaccurate measurements, while reproducible, remain scientifically useless without reference to ground truth, highlighting the need for well-established 'gold standards' in experimental measurements [82].
The Distinction Between Cancer-Initiating and Cancer-Propagating Cells
In oncogenic potential research, clarifying terminology is essential for proper experimental design and interpretation. The cell-of-origin of cancer (cancer-initiating cell) is the normal cell that receives the first cancer-causing mutations, while cancer stem cells (CSCs) (cancer-propagating cells) maintain tumor propagation through self-renewal and multipotency [83]. These represent distinct cell populations with different functional properties, though they are often mistakenly used interchangeably [83].
Figure 1: Relationship between normal cells, cell-of-origin, and cancer stem cells in tumor development.
Standardized Assay Approaches for Oncogenic Potential Assessment
Transplantation Assays: The Traditional Gold Standard
The serial tumor transplantation assay represents the current gold standard for identifying CSCs, as it directly assesses both self-renewal and multipotency—the defining properties of stem cells [83]. In this approach, tumor cell populations are fractionated using fluorescence-activated cell sorting (FACS) based on putative CSC markers, followed by limiting dilution assay (LDA) and serial transplantation into immunocompromised mice [83]. Populations that give rise to serially transplantable tumors that histologically recapitulate the cellular heterogeneity of the parental tumors are classified as CSCs [83].
The key elements for proper implementation include rigorous LDA to measure CSC frequency and serial transplantations to confirm self-renewal capacity in vivo [83]. For example, in prostate cancer, PSA-/lo human prostate cancer cells could regenerate and propagate xenograft tumors virtually indefinitely, whereas PSA+ cells could only propagate tumors for approximately three generations [83]. Transplantation assays can also probe the potential cell-of-origin of cancer by introducing oncogenic events into sorted normal cell subpopulations and assessing their tumor-forming capacity [83].
However, transplantation assays have significant limitations. Dissociated single cells may not behave the same way as in their natural tissue microenvironment, potentially misrepresenting the existence or abundance of CSCs [83]. Additionally, the lack of an immune-competent microenvironment in immunodeficient mice means these assays may not fully recapitulate the in vivo situation where immune surveillance plays a crucial role in tumor development [83].
Lineage Tracing: Assessing Cellular Potential in Native Environments
Lineage tracing has emerged as the gold standard for defining the cell-of-origin of transformation in mouse models, providing insight into the proliferative potential and fate of stem cells during tumor formation within their native microenvironment [83]. This approach allows researchers to follow the progeny of specific marked cells over time, enabling the determination of clonal relationships and differentiation trajectories without disrupting the tissue architecture [83].
The major advantage of lineage tracing over transplantation assays is its ability to study cellular behavior in unperturbed tissue contexts, maintaining natural cell-cell interactions, signaling gradients, and tissue architecture that influence stem cell function [83]. When combined with inducible Cre-lox systems, lineage tracing enables temporal control over oncogene activation or tumor suppressor inactivation, allowing researchers to precisely track the contribution of specific cell populations to tumor initiation and maintenance [83].
Molecular Detection Methods: Glycan-Based CSC Identification
Novel approaches for CSC detection have emerged that exploit specific glycosylation patterns on the cell surface. Researchers have developed a method using a combination of biotinylated plant lectins (UEA-1 and GSL-I) that selectively recognize glycan patterns expressed exclusively by CSCs [84]. This approach, commercially known as LungSTEM for non-small cell lung cancer (NSCLC), has demonstrated superior CSC detection capabilities compared to traditional markers like CD133 [84].
In validation studies, the lectin MIX+ sorted fraction showed significant CSC enrichment compared to CD133+ cells and demonstrated high tumorigenic capacity in vivo [84]. Importantly, this method has proven clinical significance, with its prognostic value for overall survival in early-stage NSCLC patients validated in a cohort of 221 patients [84]. The approach enables detection of CSCs directly linked to tumor aggressiveness and could help identify patients at high risk of relapse [84].
Comparative Analysis of Assay Performance and Standardization
Quantitative Comparison of CSC Detection Methods
Table 1: Performance comparison of different cancer stem cell detection methods
| Method | Key Metrics | Advantages | Limitations | Reproducibility Concerns |
|---|---|---|---|---|
| Transplantation Assays | CSC frequency (LDA); Serial transplantation capacity; Multilineage differentiation | Gold standard for functional validation; Assesses self-renewal and differentiation in vivo | Does not preserve native microenvironment; Immune-deficient hosts don't mimic human immunity; Labor-intensive and time-consuming | Frequency varies with level of immunodeficiency (1-25% in different mouse models); Influenced by tumor malignancy level [83] |
| Lineage Tracing | Clonal contribution to tumor; Differentiation patterns; Cell fate mapping | Studies cells in native microenvironment; No disruption of tissue architecture; Temporal control of oncogenic events | Limited to genetically engineered mouse models; Cannot be applied to human patients directly; Technical complexity of tracing systems | High reproducibility in controlled genetic models; Less variable than transplantation between laboratories |
| Lectin-Based Detection (LungSTEM) | CSC enrichment factor; Sphere formation efficiency; Tumorigenic potential in vivo; Prognostic value for survival | Directly applicable to human samples; Compatible with clinical techniques (IHC); Higher specificity for CSCs than CD133 | Limited to specific cancer types currently; Requires validation for each new cancer type; Glycosylation patterns may vary | Clinical validation on 221 patients; Significant prognostic value for OS [84] |
| CD133-Based Detection | Percentage of CD133+ cells; Sphere formation; In vivo tumor initiation | Well-established marker across multiple cancers; Extensive literature for comparison | Lacks specificity as CD133 protein present in both CSCs and differentiated cells [84]; Epitope masking affects detection | Poor prognostic value compared to lectin method; Inconsistent correlation with clinical outcomes [84] |
Standardization Approaches Across Model Systems
The International Brain Laboratory (IBL) provides an exemplary case study in standardization for complex biological measurements across multiple laboratories [85]. By implementing standardized training protocols, experimental hardware, software, and procedures for measuring decision-making in mice, they achieved reproducible results across seven laboratories in three countries [85]. Their approach included:
- Standardized variables: Mouse strain and provider, age range, weight range, water access, food protein, and fat content [85]
- Documented but not standardized variables: Light-dark cycle, temperature, humidity, and environmental sound [85]
- Open science approach: Sharing all hardware and software components, experimental protocols, and data through an open-access pipeline [85]
This systematic standardization enabled the collection of 5 million mouse choices into a publicly available database with reproducible behavioral measurements despite differences in learning speed across laboratories [85].
Detailed Experimental Protocols
Lectin-Based CSC Detection and Sorting Protocol
The following protocol adapts the method validated for detecting lung CSCs using a specific combination of lectins (UEA-1 and GSL-I) [84]:
Sample Preparation: Create a single-cell suspension at a concentration of 2×10^7 cells/mL in appropriate buffer. Exclude dying cells by adding propidium iodide (PI) to the suspension.
Staining Procedure: Incubate cells with the biotinylated lectin MIX (at the specific ratio described in patents FR20170055137 and FR20170055139) for 30 minutes at 4°C.
Cell Sorting: Sort labeled cells using FACS (BD FACSAria III sorters recommended for single-cell sorting) based on lectin fluorescence intensity. Include appropriate controls (unstained cells, single lectin stains) for gating optimization.
Validation Assays:
- Clonogenic assay: Plate sorted cells in ultra-low attachment 96-well plates at decreasing cell densities (1000, 100, 10, and 1 cell per well) in defined 3D culture medium. Quantify sphere formation weekly for 4-8 weeks.
- In vivo tumorigenesis: Inject sorted cell fractions into immunocompromised mice (e.g., NSG) at limiting dilutions to assess tumor-initiating capacity.
- Molecular characterization: Analyze sorted populations for stemness markers (Nanog, Oct4, Sox2) using RT-qPCR.
Clinical Correlation: For prognostic validation, apply the lectin staining to patient tumor sections and correlate staining intensity with clinical outcomes (overall survival, relapse-free survival).
Molecular Profiling Tests for Breast Cancer Stratification
Table 2: Comparison of genomic assays for early-stage breast cancer stratification
| Assay Name | Genes Analyzed | Technology | Clinical Utility | Predictive/Prognostic Evidence |
|---|---|---|---|---|
| Oncotype DX | 21 genes (16 cancer + 5 reference) | Quantitative RT-PCR | Predicts chemotherapy benefit in node-negative and node-positive (1-3 nodes) HR+/HER2- breast cancer | Level 1 evidence from TAILORx (node-negative) and RxPONDER (node-positive) trials [86] [87] |
| MammaPrint | 70 genes | Microarray | Prognostic stratification for early-stage breast cancer | FDA-cleared; prognostic validation in multiple studies [87] |
| Prosigna (PAM50) | 50 genes | nCounter-based gene expression | Risk of recurrence classification; intrinsic subtyping | FDA-cleared; prognostic in hormone receptor-positive disease [88] |
| Breast Cancer Index | 7 genes | RT-PCR | Predicts late recurrence and benefit from extended endocrine therapy | Unique predictive value for extended therapy decisions [87] |
| EndoPredict | 12 genes | RT-PCR | Risk stratification for early-stage ER+ breast cancer | Prognostic validation; can be combined with clinical parameters [87] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key research reagents and their applications in oncogenic potential assays
| Reagent/Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Cell Surface Markers | CD133, CD44, CD24, EpCAM | Identification and isolation of putative cancer stem cell populations | Lack specificity individually; often used in combination; epitope masking concerns with CD133 [84] |
| Lectin Combinations | UEA-1, GSL-I (LungSTEM MIX) | Detection of CSC-specific glycan patterns on cell surface | Higher specificity for functional CSCs than conventional markers; validated in clinical cohorts [84] |
| Stemness Transcription Factors | Antibodies against Nanog, Oct4, Sox2 | Identification of stem cell molecular signature | Nuclear localization requires cell fixation/permeabilization; not suitable for live cell sorting |
| 3D Culture Matrices | Matrigel, Ultra-low attachment plates | Maintenance of stem cell properties in vitro | Preserves stemness better than 2D culture; enables sphere formation assays [84] |
| Cytometry Reagents | Propidium iodide, viability dyes | Exclusion of dead cells during sorting | Critical for accurate quantification; dead cells can non-specifically bind antibodies and lectins |
| Molecular Profiling Kits | qRT-PCR reagents, RNA extraction kits | Gene expression analysis of stemness pathways | Standardized protocols essential for reproducibility; housekeeping gene selection critical |
Signaling Pathways and Experimental Workflows in Oncogenic Potential Assessment
Figure 2: Integrated experimental workflow for assessing oncogenic potential across stem cell types.
Overcoming technical variability in assays for oncogenic potential assessment requires a multifaceted approach that addresses standardization at every experimental stage. The convergence of functional assays (transplantation, lineage tracing) with molecular detection methods (lectin-based profiling, genomic tests) provides complementary information that enhances the reliability of CSC identification and characterization [83] [84].
Critical to improving reproducibility is the implementation of standardized protocols, rigorous validation across multiple laboratories, and open science practices that make data, code, and methodologies freely available [82] [85]. As the field moves forward, leveraging the strengths of different assay systems while acknowledging their limitations will be essential for generating reproducible, clinically relevant insights into cancer initiation and progression across different stem cell types.
The transformative potential of human induced pluripotent stem cells (iPSCs) in regenerative medicine is concurrently challenged by two significant safety hurdles: tumorigenic risk from residual undifferentiated cells and genomic instability acquired during reprogramming. These interlinked risks represent a formidable obstacle to the clinical implementation of iPSC-derived therapies, as they directly influence the oncogenic potential of the final cell product [89]. Teratoma formation, a risk posed by pluripotent cells that escape purification processes, necessitates rigorous detection and elimination strategies. Simultaneously, the very process of reprogramming somatic cells to a pluripotent state can introduce genomic alterations that may confer selective growth advantages or predispose cells to malignant transformation [90] [91]. This objective analysis compares current methodologies for quantifying, understanding, and mitigating these iPSC-specific risks, providing researchers with experimental data and protocols essential for comprehensive oncogenic risk assessment.
Teratoma Formation: Mechanisms and Detection Strategies
Understanding the Teratoma Risk
Teratoma formation risk originates from residual undifferentiated human pluripotent stem cells (hPSCs) that may persist in cell therapy products (CTPs). These cells can form benign growths containing tissues from all three germ layers, a direct consequence of their pluripotent nature [92]. The risk is not merely theoretical; even a small number of residual undifferentiated cells can potentially lead to tumor formation upon transplantation. The "Cancer Stem Cell" concept, which has evolved since the 19th century, provides a relevant framework for understanding how small, potent cell populations can drive tumor initiation and growth, mirroring concerns with residual iPSCs [8]. The challenge is amplified by the dynamic nature of these cells, which can adapt and evade detection, making comprehensive risk assessment protocols essential for clinical safety [92].
Experimental Detection and Quantification Methods
Robust detection of residual pluripotent cells is critical for quality control. The table below summarizes key in vitro and in vivo assays used to evaluate teratoma formation risk.
Table 1: Assays for Teratoma Risk Assessment of iPSC-Derived Products
| Method Category | Specific Assay | Measured Endpoint | Detection Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| In Vitro Pluripotency Assays | Soft Agar Colony Formation (SACF) | Colony formation in non-adherent conditions | Moderate | Measures transformation potential; quantifiable | May not detect all tumorigenic cells |
| Growth in Low Attachment (GILA) | Colony formation capacity | Varies with method | Simple setup; no animal required | Limited physiological relevance | |
| In Vivo Tumorigenicity | Immunodeficient Mouse Models (e.g., NSG, NOG) | Teratoma formation incidence and timeline | High (can detect <10^6 cells) | Gold standard; provides physiological context | Time-consuming (6-12 weeks); costly; ethical considerations |
| Molecular Detection | qPCR/dPCR for Pluripotency Markers | Expression of OCT4, SOX2, NANOG | Very High (detects 1 in 10^5-10^6 cells) | Highly sensitive; quantitative; rapid | Does not confirm functional tumorigenicity |
| Flow Cytometry with Lectins (rBC2LCN) | Surface marker expression (e.g., TRA-1-60, SSEA-4) | Moderate to High | Can detect single cells; relatively fast | Marker expression may vary with culture conditions | |
| Next-Generation Sequencing | RNA-seq for Pluripotency Signatures | Transcriptomic profiles | High | Comprehensive; can detect subtle changes | Complex data analysis; higher cost |
Recent consensus recommendations from the Health and Environmental Sciences Institute emphasize that detection sensitivity must be calibrated to the specific clinical application, with more sensitive methods required for products with systemic delivery routes [92]. Quantitative PCR (qPCR) and digital PCR (dPCR) assays targeting pluripotency-associated genes like OCT4 and SOX2 can achieve sensitivities sufficient to detect one undifferentiated cell among 10^5 to 10^6 differentiated cells [92]. For functional assessment, the tumor-producing dose at the 50% end-point (TPD50) in severe combined immunodeficiency (SCID) mice remains a critical, though resource-intensive, benchmark [92].
Experimental Protocol: In Vivo Teratoma Assay
Purpose: To assess the tumorigenic potential of iPSC-derived cell products in vivo. Materials: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, 4-6 weeks old; Test cell product (minimum 1×10^6 cells); Matrigel matrix; PBS. Procedure:
- Cell Preparation: Harvest iPSC-derived product and resuspend in 50:50 PBS:Matrigel mixture. Keep on ice.
- Injection: Subcutaneously inject 2×10^6 cells in 100μL total volume into the dorsal flank of anesthetized NSG mice [91].
- Monitoring: Palpate weekly for tumor formation over 12-16 weeks. Measure tumor dimensions with calipers.
- Endpoint Analysis: Excise tumors, fix in 4% formaldehyde, and process for H&E staining. Histologically examine for tissues from all three germ layers. Interpretation: The assay is positive if well-differentiated teratoma structures are observed. The time to tumor formation and incidence rate provide comparative risk assessment between different cell products or manufacturing processes.
Diagram: Teratoma Risk Assessment Pathway. This workflow compares in vivo and in vitro methods for detecting tumorigenic residual iPSCs.
Genomic Instability in iPSC Reprogramming and Culture
The reprogramming of somatic cells to pluripotency imposes significant stress that can lead to various genomic alterations. Recent systematic investigations have revealed that copy number alterations (CNAs) and single-nucleotide variations (SNVs) can arise during reprogramming, differentiation, and passaging phases [91]. The choice of reprogramming method significantly impacts genomic stability, with Sendai virus (SV) methods showing a higher frequency of CNAs compared to episomal vector (Epi) approaches. Specifically, studies found that 100% of SV-iPS cell lines exhibited CNAs during reprogramming, while only 40% of Epi-iPS cells showed such alterations [91]. Furthermore, SNVs were observed exclusively in SV-derived cells during passaging and differentiation, highlighting method-dependent vulnerability.
The TP53 tumor suppressor pathway is particularly vulnerable during reprogramming. TP53 mutations are frequently identified in iPSCs, underscoring the critical need for careful genomic scrutiny when preparing iPS cells and derived cell lines [91]. Additionally, prolonged culture exacerbates these issues, with late-passage cells showing upregulation of chromosomal instability-related genes, further compounding the oncogenic risk profile of iPSC-derived products.
Comparative Analysis of Reprogramming Methods
Table 2: Genomic Instability Profiles Across Reprogramming Methods
| Reprogramming Method | Copy Number Alterations (CNAs) | Single Nucleotide Variations (SNVs) | Integration Risk | Relative Genomic Stability | Key Applications |
|---|---|---|---|---|---|
| Sendai Virus (SV) | High (100% of lines in studies) | Present in passaging/differentiation | Non-integrating | Lower | Research, clinical with stringent QC |
| Episomal Vectors (Epi) | Moderate (40% of lines) | Not detected in studies | Non-integrating | Higher | Clinical applications preferred |
| mRNA Transfection | Low to Moderate | Minimal reported | Non-integrating | High | Clinical-grade iPSC generation |
| Integrating Viral (Retro/Lenti) | High | Elevated | Integrating | Lowest (unsafe for clinical use) | Basic research only |
The data reveal clear trade-offs between reprogramming efficiency and genomic integrity. While Sendai virus systems offer efficient reprogramming, they impose greater genomic stress. In contrast, episomal vectors provide enhanced genomic stability but may have lower efficiency in some cell types [91]. Recent advances in non-integrative methods, including mRNA transfection and small molecule-based reprogramming, further reduce genome-changing risks during reprogramming by avoiding integration entirely and enabling more controlled transgene expression [90].
Experimental Protocol: Comprehensive Genomic Instability Screening
Purpose: To identify and quantify genomic alterations in iPSC lines and their derivatives. Materials: High-quality DNA from iPSCs (>50ng/μL); SNP microarray or whole-genome sequencing platform; Next-generation sequencing system; Bioinformatics analysis pipeline. Procedure:
- Sample Collection: Harvest iPSCs at early (P10) and late (P30) passages, plus differentiated cell products.
- Chromosomal Microarray Analysis: Hybridize DNA to SNP arrays to detect CNAs and loss of heterozygosity.
- Next-Generation Sequencing: Perform whole-exome or whole-genome sequencing (30-50x coverage) to identify SNVs and small indels.
- Bioinformatics Analysis:
- Align sequences to reference genome (GRCh38)
- Call variants using GATK best practices
- Filter against population databases (gnomAD) to identify novel variants
- Annotate variants for functional impact
- Validation: Confirm high-priority variants by Sanger sequencing. Interpretation: Focus on mutations in cancer-associated genes (e.g., TP53), large CNAs (>1Mb), and variants absent in parental somatic cells. Establish thresholds for acceptable genomic variation based on intended clinical use.
Diagram: Genomic Instability Pathways in iPSC Generation. This chart shows how reprogramming methods and culture duration contribute to genetic alterations.
Emerging Technologies for Risk Mitigation
Advanced Detection and Prevention Strategies
The field is rapidly evolving beyond basic detection methods toward more sophisticated prevention and elimination strategies. Artificial intelligence (AI) approaches are demonstrating remarkable potential in identifying stemness characteristics and predicting tumorigenic risk. Machine learning tools can now process multi-omics data to define stemness signatures and identify rare CSC populations, with applications in iPSC quality control [71]. For instance, the CANDiT (Cancer Associated Nodes for Differentiation Targeting) AI platform can identify treatment targets that reprogram cancer stem cells to self-destruct, a approach that could be adapted for eliminating residual iPSCs [93].
CRISPR-based technologies are also contributing to safety advances. AI models for CRISPR guide RNA design are enhancing the precision of genome editing, improving both on-target efficiency and off-target safety predictions [94]. These tools enable more accurate genetic correction of patient-specific iPSCs while minimizing unintended mutations that could increase oncogenic risk [90]. Furthermore, base editors and prime editors enable exact gene modification with fewer errors while avoiding double-strand break formation, thereby minimizing unintended mutations [90].
For direct elimination of tumorigenic cells, current strategies primarily target hPSC-specific surface markers or physiological properties. These include:
- Antibody-based cytotoxicity: Targeting specific cell surface markers (e.g., TRA-1-60, SSEA-4) expressed on pluripotent cells
- Metabolic selection: Exploiting differential sensitivity to drugs between pluripotent and differentiated cells
- Physical separation: Using size, density, or adhesion properties to enrich for differentiated populations
The Scientist's Toolkit: Essential Reagents for iPSC Risk Assessment
Table 3: Key Research Reagents for iPSC Risk Assessment
| Reagent/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Pluripotency Markers | Anti-OCT4, SOX2, NANOG antibodies | Immunodetection of residual pluripotent cells | Flow cytometry, immunocytochemistry |
| Cell Surface Markers | TRA-1-60, SSEA-4, rBC2LCN lectin | Live cell identification and sorting | FACS, magnetic bead separation |
| Reprogramming Kits | CytoTune-iPS 2.0 Sendai, Episomal Vectors | Footprint-free iPSC generation | iPSC line derivation |
| Genomic Analysis | SNP Microarray Kits, Whole Genome Sequencing | Detection of CNAs and SNVs | Genomic stability assessment |
| In Vivo Models | NSG, NOG mice | Teratoma formation assays | Functional tumorigenicity testing |
| AI/Computational Tools | CANDiT, CRISPRon, Stemness Index | Predictive analysis of tumorigenic risk | Computational risk assessment |
Comparative Risk Assessment and Future Directions
When evaluating the relative challenges in iPSC safety, teratoma formation and genomic instability present distinct yet interconnected risk profiles. Teratoma risk can be mitigated through rigorous purification and detection protocols, with current sensitive assays capable of detecting rare undifferentiated cells. In contrast, genomic instability presents a more complex challenge, as mutations may not manifest immediately but could predispose cells to malignant transformation long after transplantation.
The emerging paradigm integrates multiple risk mitigation strategies:
- Reprogramming Method Selection: Prioritizing non-integrating methods with superior genomic stability profiles, such as episomal vectors or mRNA transfection [91]
- Comprehensive Genomic Screening: Implementing orthogonal methods (microarray + NGS) at multiple manufacturing stages
- Advanced Purification: Leveraging surface markers and metabolic selection to deplete residual pluripotent cells
- Functional Validation: Employing sensitive in vivo models to confirm tumorigenic potential
- Computational Prediction: Utilizing AI tools to identify high-risk signatures and optimize differentiation protocols
The field is moving toward increasingly sophisticated quality control frameworks that address both teratoma risk and genomic instability in an integrated manner. As these technologies mature, they promise to enhance the safety profile of iPSC-derived therapies, accelerating their translation to clinical applications while maintaining rigorous safety standards essential for regenerative medicine.
The integration of rigorous tumorigenicity assessment into the manufacturing processes of stem cell-based therapies is a critical frontier in regenerative medicine and oncology. As these advanced therapies move from research to clinical application, a comprehensive biosafety evaluation is essential to ensure patient safety, particularly concerning risks of oncogenicity, tumorigenicity, and teratogenicity [7]. Tumorigenicity—the potential for cells to form tumors upon transplantation—presents a complex challenge that varies significantly across different stem cell types, including induced pluripotent stem cells (iPSCs), embryonic stem cells (ESCs), mesenchymal stem/stromal cells (MSCs), and engineered cell derivatives [7] [95]. A risk-based approach, tailored to the specific cell type, its manipulation, and its intended clinical use, is paramount for developing effective quality control pipelines [96]. This guide objectively compares current methodologies, experimental protocols, and technological solutions for integrating these assessments, providing a framework for researchers and drug development professionals operating within the broader context of oncogenic potential assessment across stem cell types.
Comparative Analysis of Tumorigenicity Risks Across Stem Cell Types
The oncogenic potential of stem cell products is not uniform; it is intrinsically linked to the cell's origin, differentiation status, and manufacturing history. Understanding these differences is the first step in designing a targeted quality control pipeline.
Table 1: Comparative Tumorigenicity Profiles of Major Stem Cell Types
| Stem Cell Type | Key Tumorigenicity Risks | Primary Risk Factors | Recommended Core Assessment Methods |
|---|---|---|---|
| Induced Pluripotent Stem Cells (iPSCs) | Teratoma formation, genetic instability from reprogramming, tumor formation from residual undifferentiated cells [7] [95]. | Reactivation of reprogramming factors, mutational load acquired during reprogramming and expansion, incomplete differentiation [96]. | Karyotyping, whole-genome sequencing, in vivo teratoma assay, in vitro pluripotency tests (e.g., flow cytometry for pluripotency markers). |
| Embryonic Stem Cells (ESCs) | Teratoma formation, genetic and epigenetic alterations from long-term culture [95]. | Similar to iPSCs; risk of residual undifferentiated cells in the final product. | Same as for iPSCs, with particular emphasis on genetic stability over long-term culture. |
| Mesenchymal Stem/Stromal Cells (MSCs) | Ectopic tissue formation, spontaneous malignant transformation in vitro, supporting existing tumor growth [7]. | Donor age and tissue source, high in vitro passage number, culture conditions, interaction with the tumor microenvironment [7] [97]. | Karyotyping, soft agar colony formation assay, in vivo tumor formation in immunocompromised models, assessment of senescence. |
| Engineered Cell Therapies (e.g., CAR-T, iPSC-derived) | Insertional oncogenesis, off-target effects of gene editing, uncontrolled proliferation of engineered cells [7] [96]. | Vector design (for viral transduction), specificity of gene-editing nucleases (e.g., CRISPR-Cas9), potency and persistence of the product. | Integration site analysis (e.g., LAM-PCR for viral vectors), off-target mutation assessment (e.g., GUIDE-seq for CRISPR), in vivo biodistribution and tumorigenicity studies. |
Integrated Quality Control Pipeline: From Cell Bank to Final Product
A state-of-the-art quality control pipeline for tumorigenicity must be a multi-stage, risk-based process integrated throughout manufacturing. The following workflow diagram and subsequent text outline this continuous assessment strategy.
Diagram 1: Integrated tumorigenicity assessment workflow.
Critical Stages of the Integrated Pipeline
Master Cell Bank (MCB) Characterization: This is the most critical stage for risk reduction. A fully characterized, clonally derived MCB reduces inherent risks for iPSC-derived allogeneic therapies, as genetic engineering and donor selection are performed once [96]. Comprehensive profiling here includes whole-genome sequencing to identify oncogenic mutations, karyotyping for chromosomal abnormalities, and in vitro transformation assays.
In-Process Controls: Monitoring during manufacturing, especially for processes involving differentiation, expansion, and genetic modification, is essential. This includes checks for genetic stability (e.g., via SNP arrays), removal of undifferentiated cells, and confirmation of the target phenotype. For genetically modified cells, this stage should include analyses for vector copy number and editing efficiency.
Final Product Release Testing: The final product must pass stringent release criteria, which are defined based on the risk assessment from earlier stages. Key assays include:
- Sterility and Viability: Ensuring the product is free from microbial contamination and has appropriate viability [7].
- Identity and Purity: Confirming the product is the intended cell type and assessing the level of residual undifferentiated cells (e.g., via flow cytometry for pluripotency markers) [7].
- Potency: Demonstrating the biological function of the product, which is often linked to its therapeutic mechanism [7].
- Genetic Stability: Repeating critical genetic tests (e.g., karyotyping) to confirm stability through the manufacturing process.
Experimental Protocols for Key Tumorigenicity Assays
This section provides detailed methodologies for the core experiments used to assess tumorigenic potential, forming the backbone of the quality control pipeline.
In Vivo Tumorigenicity and Teratoma Assay
The in vivo assay is considered the gold standard for evaluating a cell product's potential to form tumors or teratomas in a living organism [7].
- Objective: To determine the capacity of the stem cell product to form undesired growths, including teratomas (for pluripotent cells) or tumors, upon implantation into an immunocompromised host.
- Materials:
- Test Cells: Final product cell population or cells from the MCB.
- Control Cells: Known tumorigenic cell line (e.g., HeLa) as a positive control, and a non-tumorigenic cell line as a negative control.
- Animals: Immunocompromised mice (e.g., NOD-scid gamma (NSG) or nude mice) [7].
- Equipment: Cell culture hood, injector, in vivo imaging system (optional).
- Procedure:
- Cell Preparation: Harvest and resuspend test and control cells in an appropriate, non-immunogenic matrix like Matrigel or PBS.
- Implantation: Inject a range of cell doses (e.g., from 1x10^6 to 1x10^7 cells) into the intended clinical delivery site (e.g., subcutaneous, intramuscular, or renal capsule). For teratoma assays with pluripotent cells, the renal capsule or testis are sensitive sites.
- Observation and Monitoring: Monitor animals for a sufficient period (e.g., 12-20 weeks) for palpable mass formation or signs of ill health. Use in vivo imaging if cells are labeled.
- Necropsy and Histopathology: Euthanize animals at the study endpoint or if they show adverse effects. Excise and weigh any masses. Perform a comprehensive histological examination (H&E staining) of masses and key organs (liver, lungs, kidneys) to identify tissue types in teratomas or malignant features in tumors [7].
- Data Interpretation: The formation of a mass containing cells from all three germ layers (ectoderm, mesoderm, endoderm) confirms teratoma formation. The incidence and latency of tumor formation at different cell doses inform the product's tumorigenic risk.
In Vitro Soft Agar Colony Formation Assay
This assay measures anchorage-independent growth, a hallmark of cellular transformation that strongly correlates with in vivo tumorigenicity.
- Objective: To assess the transformation potential of stem cells by quantifying their ability to form colonies in a semi-solid medium without attachment.
- Materials:
- Agarose
- Base Agar Layer: Culture medium with 0.5% - 1.0% agarose.
- Top Agar Layer: Culture medium with 0.3% - 0.5% agarose containing the suspended test cells.
- Control Cells: As in the in vivo assay.
- Procedure:
- Base Layer Preparation: Pour the base agar layer into multi-well plates and allow it to solidify.
- Cell Seeding: Mix the test and control cells with the top agar layer and plate it over the solidified base layer.
- Incubation and Feeding: Culture the plates for 3-4 weeks, adding fresh culture medium weekly to prevent drying.
- Staining and Counting: Stain the plates with a viability dye like INT or MTT. Count the number and size of colonies using an automated colony counter or microscope.
- Data Interpretation: A statistically significant increase in the number and size of colonies formed by the test cells compared to the non-tumorigenic negative control indicates transformation potential.
Table 2: Summary of Key Tumorigenicity Assays and Their Applications
| Assay Type | Measured Endpoint | Duration | Key Advantages | Key Limitations |
|---|---|---|---|---|
| In Vivo Tumor Formation | Actual tumor/teratoma formation in a live model [7]. | 12-20 weeks | Provides a holistic, physiological assessment; gold standard. | Time-consuming, expensive, low-throughput, ethical considerations. |
| Soft Agar Colony Formation | Anchorage-independent growth (transformation) [7]. | 3-4 weeks | Quantitative, relatively low-cost, high-throughput potential. | Does not fully recapitulate the in vivo tumor microenvironment; may produce false negatives/positives. |
| Karyotyping / Genomic Sequencing | Genetic stability, chromosomal abnormalities, oncogenic mutations [7] [96]. | 1-4 weeks | Identifies root causes of tumorigenicity; high specificity. | May miss epigenetically driven tumorigenicity; does not assess functional outcome. |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following reagents and tools are fundamental for establishing a robust tumorigenicity assessment pipeline.
Table 3: Key Research Reagents for Tumorigenicity Assessment
| Reagent / Solution | Function in Assessment Pipeline |
|---|---|
| Immunocompromised Mouse Models (e.g., NSG, Nude) | In vivo hosts for tumorigenicity and teratoma assays, allowing the growth of human cells without immune rejection [7]. |
| Extracellular Matrix (e.g., Matrigel) | Provides a supportive 3D environment for cell implantation in in vivo assays and for more physiologically relevant in vitro 3D culture models [98]. |
| Pluripotency Marker Antibodies (e.g., anti-SOX2, OCT4, NANOG) | Critical for flow cytometry and immunohistochemistry to assess the purity of differentiated products and identify residual undifferentiated cells [8] [95]. |
| Next-Generation Sequencing (NGS) Kits | For comprehensive genomic profiling, including whole-genome sequencing to detect mutations and off-target effects of gene editing, and single-cell RNA sequencing to characterize heterogeneity [95]. |
| Microphysiological Systems (Organ-on-a-Chip) | Advanced in vitro models that mimic the human tumor microenvironment, allowing for the study of cell-cell interactions and tumorigenic potential in a more human-relevant system [97]. |
Emerging Technologies and Future Perspectives
The field of tumorigenicity assessment is being transformed by the integration of next-generation technologies that promise enhanced precision, throughput, and human relevance.
- Advanced In Vitro Models (MPS): Microphysiological systems, or organs-on-chips, are revolutionizing biosafety assessment. These microfluidic devices can replicate key features of the in vivo microenvironment, such as physiological flow, shear stress, and multi-cellular interactions, providing a more predictive platform for evaluating tumorigenic potential than traditional 2D cultures [97]. When combined with bioprinting and real-time biosensing, MPS can dissect the complex role of the tumor microenvironment in promoting or suppressing tumorigenicity [97].
- Single-Cell and Spatial Multi-Omics: The application of single-cell RNA sequencing and spatial transcriptomics allows for the unprecedented resolution of intra-tumoral heterogeneity [8]. These technologies can identify rare, potentially tumorigenic subpopulations within a manufactured cell product that might be missed by bulk analyses, enabling a more sensitive assessment of risk [95].
- AI-Driven Risk Prediction: The integration of large-scale genomic, transcriptomic, and functional data with machine learning algorithms is paving the way for computational models that can predict the tumorigenic risk of a cell product based on its molecular signature, potentially reducing the reliance on lengthy in vivo studies in the future [8].
Benchmarking Safety: Comparative Analysis and Regulatory Validation Frameworks
The therapeutic potential of stem cells in regenerative medicine and oncology is vast, yet their clinical application is balanced by a critical assessment of their oncogenic risk. This risk—the potential to initiate or promote cancer—varies significantly between different stem cell types and is a primary consideration for researchers and drug development professionals. Pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), possess the hallmark abilities of unlimited self-renewal and differentiation into any cell type, which inherently raises concerns about teratoma formation and uncontrolled growth [99]. In contrast, adult stem cells (e.g., mesenchymal, hematopoietic), being multipotent with more limited differentiation fates, are generally considered to have lower intrinsic oncogenic potential, though they present distinct risks such as malignant transformation during extensive in vitro expansion and potential tumor-supporting roles through paracrine signaling [22] [95]. This guide provides a objective, data-driven comparison of the oncogenic risks associated with PSCs and adult stem cells, framing the discussion within the broader thesis of risk assessment for therapeutic development.
Comparative Oncogenic Risk Profiles
The oncogenic potential of stem cells is governed by their origin, biological properties, and response to manipulation. The table below summarizes the core risk profiles and associated mechanisms for the two main classes.
Table 1: Fundamental Oncogenic Risk Profiles of Major Stem Cell Types
| Stem Cell Type | Key Defining Characteristics | Primary Oncogenic Risks | Underlying Mechanisms & Contributing Factors |
|---|---|---|---|
| Pluripotent Stem Cells (PSCs)(e.g., ESCs, iPSCs) | - Pluripotency (can form all germ layers)- Virtually unlimited self-renewal capacity in vitro [99] | - Teratoma formation post-transplantation- Incomplete differentiation leading to persistent proliferative cells- Genomic instability in iPSCs due to reprogramming [95] | - Presence of undifferentiated cells in final product- p53 pathway suppression during reprogramming- Copy number variations and mutations acquired during culture |
| Adult Stem Cells(e.g., MSCs, HSCs) | - Multipotency (limited to lineages of origin)- Finite lifespan in culture without transformation [99] [22] | - Malignant transformation during long-term in vitro expansion- Tumor tropism potentially supporting tumor growth and metastasis- Immunosuppressive effects potentially shielding cancers [22] [95] | - Spontaneous transformation through sequential upregulation of oncogenes (e.g., c-myc) and downregulation of tumor suppressors (e.g., p16)- Secretion of pro-angiogenic and pro-metastatic factors |
Quantitative Risk Comparison from Experimental Studies
Translating the conceptual risks into quantifiable metrics is essential for a rigorous comparative analysis. The following table compiles key experimental data from the literature, highlighting the frequency and contexts of oncogenic events.
Table 2: Quantitative Comparison of Oncogenic Events from Preclinical and Clinical Studies
| Stem Cell Type | Experimental Context / Model | Reported Oncogenic Event Frequency | Key Observations & Outcomes |
|---|---|---|---|
| PSCs (iPSCs/ESCs) | - In vitro differentiation and genomic analysis- Teratoma assay in immunodeficient mice | - Genomic instability: A significant concern; mutations can accumulate during reprogramming and prolonged culture of iPSCs [95].- Teratoma incidence: High likelihood if even a small number of undifferentiated cells are present in a graft. | - Risk is considered a major hurdle for clinical translation.- Requires stringent purification and tumorigenicity testing of final cell product. |
| MSCs | - In vitro expansion to senescence or high passages (e.g., 4-5 months in culture) [22] | - Spontaneous transformation: Observed in human adipose-derived MSCs after 4-5 months of culture, though this phenomenon is considered highly controversial and not consistently reproduced [22].- Chromosomal abnormalities: A sub-population of cells with aneuploidy and translocations can emerge with prolonged culture [22]. | - Malignant transformation is linked to culture duration and specific isolation protocols.- Conflicting study results underscore the need for standardized manufacturing. |
| MSCs | - In vivo administration in cancer models (e.g., co-implantation with colon cancer cells) [22] | - Tumor enhancement: Higher tumor incidence and elevated proliferation, angiogenesis, and metastatic ability of cancer cells in MSC-treated groups. | - Highlights the tumor-supporting potential of MSCs via paracrine signaling and microenvironment modulation. |
Key Experimental Protocols for Oncogenic Risk Assessment
To generate the data required for a cross-comparison, standardized experimental protocols are employed. Below are detailed methodologies for two critical assays used in the field.
Protocol 1: In Vitro Malignant Transformation Assay for Adult Stem Cells
This protocol is designed to assess the risk of spontaneous transformation in adult stem cells, such as MSCs, during extended culture, a key differentiator from PSCs.
- Objective: To determine if long-term in vitro culture induces malignant transformation in MSCs.
- Key Steps:
- Cell Source & Isolation: Isolate MSCs from bone marrow or adipose tissue using density gradient centrifugation. Culture adherent cells in standard media (e.g., DMEM with fetal bovine serum). Confirm identity via ISCT criteria: plastic-adherence, positive for CD105, CD73, CD90, and negative for CD45, CD34, CD14, CD19, HLA-DR [22].
- Long-Term Culture & Passaging: Continuously culture cells, passaging them at ~80% confluence. Maintain for an extended period (e.g., 4-5 months or >25 passages) while monitoring morphology and growth rates [22] [100].
- Transformation Analysis:
- Telomerase Activity: Measure using the Telomeric Repeat Amplification Protocol (TRAP) assay. Elevated activity is a potential marker of immortalization [22].
- Karyotyping: Perform G-banding chromosomal analysis at regular intervals to detect chromosomal aneuploidy and translocations [22].
- Oncogene/Tumor Suppressor Expression: Analyze expression of genes like c-MYC (often upregulated) and p16 (often downregulated) via qPCR or western blot [22].
- Interpretation: Evidence of sustained rapid proliferation, high telomerase activity, significant chromosomal abnormalities, and dysregulation of key oncogenic pathways indicates in vitro malignant transformation.
Protocol 2: In Vivo Tumorigenicity Assay for PSCs and Differentiated Derivatives
This is a cornerstone assay for evaluating the risk of teratoma or tumor formation by PSC-based products, a risk not typically assessed for adult stem cells in the same way.
- Objective: To test the capacity of a stem cell population to form tumors in vivo.
- Key Steps:
- Cell Preparation: For PSCs, use either undifferentiated cells or their differentiated progeny. A critical step is quantifying the percentage of undifferentiated cells in the final product using flow cytometry for markers like Tra-1-60 or SSEA-4.
- Animal Model: Use immunodeficient mice (e.g., NOD/SCID mice) to prevent immune rejection of human cells [8] [3].
- Cell Administration: Inject cells subcutaneously, intramuscularly, or into an organ-specific site. Common tests involve injecting varying cell doses (e.g., 10^3 to 10^6 cells) to determine tumor-initiating cell frequency [3].
- Monitoring & Endpoint Analysis:
- Palpation and Imaging: Monitor animals for tumor formation over 12-20 weeks [3].
- Necropsy and Histopathology: Upon endpoint, excise and weigh any resulting masses. Perform hematoxylin and eosin (H&E) staining to identify tissue types. For teratomas from PSCs, look for disordered tissues from all three germ layers (ectoderm, mesoderm, endoderm).
- Interpretation: Tumor formation, especially with teratoma pathology, confirms the presence of contaminating or residual pluripotent cells in a PSC-derived product. The assay validates the safety of a differentiation protocol.
Visualizing Key Signaling Pathways in Stem Cell Oncogenesis
Oncogenic transformation in stem cells often involves the dysregulation of conserved signaling pathways that control self-renewal and proliferation. The diagram below maps the core pathways and their interactions.
Figure 1: Oncogenic Signaling Pathways in Stem Cells. This map illustrates key pathways dysregulated in PSCs (red) and adult stem cells (green). PSC risk heavily involves core pluripotency factors (OCT4/SOX2/NANOG) and p53. Adult stem cell risk is linked to cell cycle regulators (p16) and telomerase, with shared pathway crosstalk (dashed lines).
The Scientist's Toolkit: Essential Research Reagents
A standardized set of reagents and tools is critical for consistent and reproducible oncogenic risk assessment across different laboratories.
Table 3: Essential Research Reagents for Oncogenic Risk Assessment
| Reagent / Tool Category | Specific Examples | Primary Function in Oncogenic Risk Assessment |
|---|---|---|
| Cell Surface Markers for Characterization & Purity | CD105, CD73, CD90 (for MSCs) [22]CD34, CD38 (for HSCs/Leukemia) [8] [3]SSEA-4, Tra-1-60 (for undifferentiated PSCs) | Definitive identification and isolation of specific stem cell populations. Critical for ensuring the absence of undifferentiated PSCs in a final product. |
| Pluripotency & Self-Renewal Pathway Modulators | CHIR99021 (WNT pathway activator) [101]DAPT (Notch pathway inhibitor) [101] | Used to manipulate signaling pathways to study their role in maintaining stemness or inducing differentiation, directly linking pathway activity to oncogenic potential. |
| In Vivo Tumorigenicity Models | NOD/SCID or NSG immunodeficient mice [8] [3] [22] | The gold-standard model for assessing the in vivo capacity of a cell population to form tumors (teratomas or other malignancies) in a permissive environment. |
| Genomic Integrity Assays | Karyotyping (G-banding) [22]Whole-genome sequencing | Detection of large-scale chromosomal abnormalities (aneuploidy, translocations) and point mutations acquired during reprogramming or long-term culture. |
A direct, quantitative comparison of oncogenic risk between PSCs and adult stem cells reveals a fundamental dichotomy: PSCs present a high, intrinsic risk of teratoma formation primarily due to their pluripotent nature, whereas adult stem cells like MSCs present a lower intrinsic risk but face extrinsic challenges of genomic instability during culture and potential tumor-supporting roles in vivo [22] [95]. For clinical translation, this necessitates distinct risk-mitigation strategies. For PSC-based therapies, the focus must be on rigorous purification of differentiated products and sensitive in vivo tumorigenicity assays. For adult stem cell therapies, the emphasis should be on establishing safe shelf-lives for cell products, monitoring genomic stability, and thoroughly investigating their impact on pre-existing or micrometastatic tumors. Future progress will rely on the integration of next-generation sequencing (NGS) for precise monitoring of genomic integrity and the development of more predictive humanized in vivo models, ultimately enabling a more precise and personalized risk-benefit analysis for stem cell-based therapies [95].
1 Introduction A foundational challenge in preclinical oncology research is the successful translation of in vitro findings to in vivo efficacy and, ultimately, to clinical outcomes. The establishment of robust correlations between laboratory models and living systems is not merely an academic exercise; it is a critical determinant of the speed, cost, and ultimate success of drug development [102] [103]. This guide objectively compares the performance of various in vitro models against in vivo benchmarks, with a specific focus on assessing the oncogenic potential of different stem cell types. We summarize key experimental data, provide detailed methodologies, and visualize core concepts to equip researchers with a practical framework for model validation.
2 The In Vitro-In Vivo Correlation (IVIVC) Framework In Vitro-In Vivo Correlation (IVIVC) is defined as a predictive mathematical model that describes the relationship between an in vitro property of a dosage form (typically the drug release rate) and a relevant in vivo response (such as plasma drug concentration or amount absorbed) [102]. The development of a robust IVIVC requires the integration of three key classes of factors:
- Physicochemical Properties: Including drug solubility, pKa, and particle size, which influence dissolution as described by the Noyes-Whitney equation [102].
- Biopharmaceutical Properties: Such as drug permeability, often estimated via octanol-water partition coefficient (logP) or polar surface area, which govern membrane transport [102].
- Physiological Properties: Including pH gradients and transit times within the gastrointestinal tract, which can alter drug stability, solubility, and permeability in vivo [102].
The U.S. Food and Drug Administration (FDA) recognizes multiple levels of IVIVC, which differ in their predictive power and regulatory utility [104]. The following table provides a comparative summary of these correlation levels.
Table 1: Levels of In Vitro-In Vivo Correlation (IVIVC)
| Aspect | Level A | Level B | Level C |
|---|---|---|---|
| Definition | A point-to-point correlation between in vitro dissolution and the in vivo absorption rate [104]. | A statistical comparison using mean in vitro dissolution time and mean in vivo residence or absorption time [104]. | A single-point relationship between one dissolution time point and one pharmacokinetic parameter (e.g., Cmax or AUC) [104]. |
| Predictive Value | High; can predict the entire plasma concentration-time profile [104]. | Moderate; does not reflect the actual shape of the in vivo profile [104]. | Low; provides only a single-point association [104]. |
| Regulatory Acceptance | Most preferred and accepted; can support biowaivers for formulation changes [104]. | Less robust; generally not sufficient for product approval or biowaivers [104]. | Least rigorous; may support early development but is insufficient for biowaivers [104]. |
The principal value of a validated IVIVC, particularly Level A, lies in its ability to serve as a surrogate for in vivo bioequivalence studies. This allows for the prediction of a drug's in vivo performance based on in vitro dissolution data, thereby reducing development timelines, minimizing the need for human and animal studies, and facilitating quality control and formulation optimization [102] [104].
3 Advanced 3D In Vitro Models for Solid Tumors Traditional two-dimensional (2D) cell cultures are limited in their ability to recapitulate the complexity of solid tumors. To address this, three-dimensional (3D) models like the Multicellular Tumor Spheroid (MCTS) have been developed as an intermediate between 2D culture and in vivo tumors [105].
Table 2: Comparison of In Vitro Tumor Models
| Model Type | Key Features | Predictive Strengths | Key Limitations |
|---|---|---|---|
| 2D Monolayer | Cells grow on a flat, plastic surface; simple and high-throughput [105]. | Rapid screening of cytotoxicity; well-established protocols [105]. | Lacks tissue structure, gradients, and cell-ECM interactions; poor clinical predictive value [105]. |
| Multicellular Tumor Spheroid (MCTS) | 3D spherical clusters of tumor cells that mimic microtumors [105]. | Reproduces proliferative, quiescent, and necrotic zones; models drug penetration gradients; better predicts treatment response [105]. | Can lack tumor microenvironment components; methods for mass production may yield heterogeneous spheroids [105]. |
| MCTS with ECM/Stromal Cells | MCTS embedded with extracellular matrix proteins and/or stromal cells (e.g., fibroblasts) [105]. | Models the physical barrier of ECM and stromal-tumor interactions; highly predictive of nanodrug penetration and efficacy [105]. | More complex and costly to establish; standardization is challenging [105]. |
The MCTS model closely mimics an avascular tumor nodule by exhibiting three fundamental characteristics also found in vivo:
- Heterogeneous Growth: The presence of an outer layer of proliferating cells, a middle layer of quiescent cells, and a central necrotic core due to nutrient and oxygen deprivation [105].
- Physiological Gradients: The establishment of oxygen, pH, and nutrient gradients from the spheroid periphery to its core [105].
- Extracellular Matrix (ECM): The presence of ECM components such as fibronectin, laminin, and collagen, which influence drug transport and tumor cell behavior [105].
These features make MCTS particularly suitable for evaluating the penetration and efficacy of novel nanotherapeutics, which rely on the Enhanced Permeability and Retention (EPR) effect for tumor targeting [105]. The workflow for establishing and using MCTS is depicted below.
4 Model Systems for Hematological Malignancies The development of physiologically relevant in vitro models for blood cancers (e.g., leukaemia, lymphoma, myeloma) has been challenging due to the complex nature of the bone marrow (BM) and lymphatic microenvironments [106]. Unlike solid tumors, hematological malignancies are systemic diseases where the tumor microenvironment (TME) plays a crucial role in disease progression and drug resistance [106] [107].
Table 3: In Vitro Models for Hematological Malignancies
| Model Type | Description | Key Applications | Considerations |
|---|---|---|---|
| 2D Co-culture | Cancer cells cultured with stromal feeder layers (e.g., Mesenchymal Stem Cells). | Studies of direct cell-cell contact and paracrine signaling; relatively simple setup [106]. | Lacks 3D architecture and physiological gradients; does not fully recapitulate the BM niche [106]. |
| 3D Scaffold-Based | Cells seeded into porous biomaterial scaffolds (e.g., polymer, alginate). | Allows for 3D cell growth and can be tailored to mimic specific ECM properties [106]. | Scaffold composition and stiffness can artificially influence cell behavior [106]. |
| Bone Marrow Organoids | 3D structures generated from iPSCs directed to differentiate into mesenchymal, endothelial, and hematopoietic lineages [107]. | Recapitulates key BM features: stroma, sinusoids, myeloid cells; supports patient-derived cell engraftment; models disease-specific fibrosis [107]. | Complex and lengthy protocol; requires specialized expertise; may not capture full heterogeneity [107]. |
A significant advancement in this field is the development of human bone marrow organoids. These organoids are generated through a directed differentiation protocol of induced pluripotent stem cells (iPSCs) and capture key features of the human BM, including:
- Stromal Components: Mesenchymal lineages that support hematopoietic cells.
- Vascular Networks: Lumen-forming sinusoids that mimic the vascular niche.
- Functional Hematopoiesis: The presence of myeloid cells, including proplatelet-forming megakaryocytes [107].
This model has been validated by its ability to support the engraftment and survival of primary cells from patients with various myeloid and lymphoid malignancies, providing a powerful ex vivo platform for target discovery and drug validation [107]. The following diagram illustrates the key signaling pathways and cellular interactions within the bone marrow niche that are modeled by these advanced systems.
5 Quantitative Correlation: From Empirical to Mechanistic Approaches In oncology, a common IVIVC approach involves correlating in vitro potency parameters (e.g., IC50 from cell proliferation assays) with the in vivo drug exposure required for efficacy, such as tumor growth inhibition in mouse xenograft models [103] [108] [109]. While empirical correlations exist, recent work focuses on developing semi-mechanistic mathematical models for a more systematic understanding.
These PK/PD/TGI (Pharmacokinetic/Pharmacodynamic/Tumor Growth Inhibition) models incorporate:
- Compound-specific parameters: In vitro IC50, Hill coefficient of the dose-response curve, and pharmacokinetic properties like peak-trough ratio (PTR).
- Xenograft-specific parameters: Tumor growth rate (g) and natural cell decay rate (d), which account for the 3D in vivo context [103] [108].
A key finding from this modeling is that for many compounds, the xenograft-specific growth and decay parameters (g and d) are more significant determinants of the efficacious dose and tumor stasis than variations in a compound's PTR. However, as the Hill coefficient (indicating cooperativity in drug effect) increases, the dependency of tumor stasis on the PTR becomes more pronounced [103] [108]. This provides a mechanistic explanation for the variability observed in empirical IVIVC and underscores the importance of characterizing both the drug and the biological system.
6 The Scientist's Toolkit: Essential Reagent Solutions The following table details key reagents and materials essential for conducting the experiments described in this guide.
Table 4: Essential Research Reagent Solutions for Oncogenic Model Validation
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| Agarose | Used to create non-adherent coatings for petri dishes in the liquid-overlay technique to promote MCTS formation [105]. | Inert and prevents cell attachment, forcing cells to aggregate. Concentration affects spheroid compactness. |
| Induced Pluripotent Stem Cells (iPSCs) | The starting cell population for generating human bone marrow organoids, capable of differentiation into all required lineages [107]. | Must be carefully characterized and maintained in a pluripotent state prior to differentiation. |
| Directed Differentiation Media | A sequence of specialized culture media containing growth factors and cytokines to direct iPSCs toward mesenchymal, endothelial, and hematopoietic fates [107]. | The precise timing and composition are critical for successful organoid development. |
| Extracellular Matrix (ECM) Hydrogels | (e.g., Matrigel, Collagen) Used to embed MCTS or cells in 3D scaffold models to provide a physiological structural and biochemical environment [105]. | Lot-to-lot variability can affect experimental reproducibility; requires optimization for specific cell types. |
| Fluorescent Cell Viability/Cytotoxicity Assays | (e.g., based on resazurin/Alamar Blue, CFSE, propidium iodide) Used to quantify cell viability and drug response in both 2D and 3D cultures [105]. | Penetration of dyes into the core of 3D models must be validated; assays may require adaptation from 2D protocols. |
| Cytokine/Antibody Panels for Flow Cytometry | Essential for immunophenotyping cells isolated from models, e.g., confirming MSC markers (CD105, CD73, CD90) or hematopoietic populations [22]. | Panels must be designed to identify and exclude contaminating cell types (e.g., hematopoietic cells from MSC cultures). |
7 Conclusion The strategic validation of in vitro models against in vivo benchmarks is paramount for de-risking the drug development pipeline. As demonstrated, the predictive value of a model increases with its physiological relevance—from simple 2D cultures to sophisticated MCTS and biomimetic bone marrow organoids. The emergence of semi-mechanistic IVIVC models provides a powerful, quantitative framework for bridging in vitro potency and in vivo efficacy, moving beyond empirical correlations. By carefully selecting and validating models that incorporate critical TME interactions and by leveraging mathematical modeling, researchers can more accurately assess oncogenic potential and therapeutic vulnerability, thereby accelerating the translation of promising discoveries from the bench to the clinic.
The cancer stem cell (CSC) hypothesis represents a paradigm shift in oncology, proposing that tumors are hierarchically organized and maintained by a subset of cells with stem-like properties [15]. These CSCs share fundamental characteristics with normal stem cells (SSCs)—primarily the capacities for self-renewal and differentiation—yet they are fundamentally different in their regulation and contribute directly to therapeutic resistance and disease relapse [110] [111]. Understanding the precise points of convergence and, more importantly, the crucial differences between normal and cancer stem cells is essential for identifying specific therapeutic windows that can be exploited for cancer treatment without damaging healthy tissues [112] [113]. This guide provides a comparative profiling of these cell types, detailing the experimental methodologies for their study and analyzing the specific biological pathways that offer the most promising targets for therapeutic intervention.
Molecular and Functional Profiling: A Side-by-Side Comparison
Core Properties and Identification Markers
The following table summarizes the key characteristics used to define and identify both normal and cancer stem cells.
Table 1: Comparative Profile of Normal Stem Cells vs. Cancer Stem Cells
| Characteristic | Normal Stem Cells (SSCs) | Cancer Stem Cells (CSCs) |
|---|---|---|
| Primary Function | Tissue homeostasis, repair, and regeneration [15] | Tumor initiation, maintenance, progression, and metastasis [84] [114] |
| Self-Renewal | Tightly regulated, often asymmetric division [111] [15] | Dysregulated, often symmetric division leading to expansion [111] |
| Proliferation Rate | Typically slow and quiescent to preserve longevity [15] | Heterogeneous, but can include rapidly proliferating subsets [111] |
| Key Surface Markers | CD34+, CD133+, CD90+ (varies by tissue) [115] | CD44+, CD133+, EpCAM+, CD90+ (varies by cancer type) [84] [114] |
| Key Intracellular Markers | Transcription factors like Oct4, Nanog, Sox2 [114] | Transcription factors like Oct4, Nanog, Sox2 [114] |
| Tumorigenic Potential | None (under normal conditions) | High; capable of forming new tumors in vivo [84] [114] |
| Therapy Resistance | Natural resistance due to quiescence and drug efflux pumps [115] | High intrinsic and acquired resistance; drivers of relapse [116] [114] |
Signaling Pathway Alterations
The dysregulation of key developmental pathways is a hallmark of CSCs. The table below compares the activity of these pathways in normal versus cancerous stem cells.
Table 2: Key Signaling Pathway Activity in Normal vs. Cancer Stem Cells
| Signaling Pathway | Role in Normal Stem Cells | Dysregulation in Cancer Stem Cells |
|---|---|---|
| Wnt/β-catenin | Regulates cell fate decisions and self-renewal in a tightly controlled manner [15] | Frequently constitutively activated, promoting uncontrolled self-renewal and tumor growth [116] [114] |
| Notch | Controls cell differentiation, proliferation, and apoptosis [15] | Can act as an oncogene or tumor suppressor depending on context; implicated in therapy resistance [116] [15] |
| Hedgehog (Hh) | Crucial for embryonic development and tissue patterning [15] | Reactivated in many cancers, contributing to CSC maintenance and proliferation [116] [15] |
| p53 | "Guardian of the genome"; induces cell cycle arrest or apoptosis in response to damage [112] [111] | Often mutated or inactivated, allowing symmetrical self-renewing divisions and genomic instability [111] |
| NF-κB | Regulates immune responses and cell survival [15] | Constitutively active, promoting a pro-inflammatory microenvironment and CSC survival [15] |
Diagram 1: Signaling Pathway Dysregulation. Key developmental pathways are tightly regulated in normal stem cells but are frequently dysregulated in CSCs, leading to unchecked self-renewal, therapy resistance, and genomic instability.
Experimental Protocols for Isolation and Characterization
Flow Cytometry-Based Isolation Methods
The isolation of CSCs and normal SSCs relies heavily on Fluorescence-Activated Cell Sorting (FACS) and Magnetic-Activated Cell Sorting (MACS). The experimental workflow, as detailed in [115] and [84], generally follows these steps:
- Single-Cell Suspension: Tumors or normal tissues are dissociated using enzymatic methods (e.g., collagenase, trypsin) to create a single-cell suspension.
- Staining and Labeling: Cells are incubated with fluorescently labeled or magnetic antibodies against specific surface markers (e.g., CD133, CD44, EpCAM). A viability dye (e.g., Propidium Iodide) is included to exclude dead cells.
- Sorting and Collection: Labeled cells are passed through a FACS or MACS system to isolate the positive and negative fractions with high purity.
- Functional Validation: The sorted populations are subjected to downstream functional assays to confirm their stem-like properties.
Diagram 2: Stem Cell Isolation Workflow. Core experimental workflow for isolating stem cell populations from tissues using surface marker expression.
Functional Assays for Stemness Evaluation
In Vivo Tumorigenicity Assay: This is the gold-standard functional test for CSCs. Serial dilutions of FACS-sorted cell populations are transplanted into immunodeficient mice (e.g., NOD/SCID). The ability to form a tumor that recapitulates the original tumor's heterogeneity upon serial transplantation is the defining feature of CSCs [115] [84] [114]. For normal SSCs, the equivalent assay is the in vivo repopulation assay, which tests the ability to reconstitute a tissue.
Sphere Formation Assay (In Vitro): Sorted cells are cultured in ultra-low attachment plates with a defined serum-free medium supplemented with growth factors (e.g., EGF, FGF, B27). The formation of non-adherent tumorspheres or mammospheres indicates the presence of self-renewing cells. This assay is applicable to both CSCs and normal SSCs [111] [84].
Side Population (SP) Assay: This functional assay is based on the high expression of ATP-binding cassette (ABC) drug transporters like ABCG2 in many stem cells. Cells are incubated with the fluorescent DNA-binding dye Hoechst 33342. Stem cells actively efflux the dye, resulting in a dimly stained "Side Population" visible by flow cytometry. The assay must include a control with an inhibitor like verapamil to confirm the specificity of the efflux [115].
The Therapeutic Window: From Concept to Clinical Application
The concept of a "therapeutic window" in CSC biology operates on two levels. First, it refers to a critical stage in the development of normal stem cells that could be harnessed for therapy, or conversely, a vulnerable stage in the conversion of a normal stem cell to a malignant CSC [112]. Second, it signifies a specific biological state or time period during which CSCs are uniquely vulnerable to targeted intervention.
Targeting the CSC "Therapeutic Window"
Several strategies are being developed to target CSCs within this therapeutic window:
- Differentiation Therapy: Forcing CSCs to differentiate into non-tumorigenic, post-mitotic cells, thereby robbing them of their self-renewal capacity and making them susceptible to conventional therapies [116].
- Targeting the CSC Niche: Disrupting the specialized microenvironment that protects and maintains CSCs. This includes inhibiting angiogenesis or specific cytokine signals [112] [15].
- Immunotherapy Approaches: Developing cancer vaccines or engineered immune cells (e.g., CAR-T) that target CSC-specific antigens to direct the immune system against the root of the tumor [116].
- Signaling Pathway Inhibitors: Using small molecule inhibitors or monoclonal antibodies to target dysregulated pathways like Wnt, Notch, and Hedgehog, which are crucial for CSC maintenance [116] [114].
Recent clinical evidence underscores the importance of this approach. A study screening over 100 FDA-approved oncology drugs found that while about half were active against CSCs, a unique subset was selective for CSCs over healthy stem cells. Crucially, these selective drugs were associated with better clinical efficacy and reduced adverse effects [117].
Table 3: Essential Research Reagents for Stem Cell Profiling
| Reagent / Tool | Primary Function | Application in Research |
|---|---|---|
| Anti-CD133/1 (AC133) | Immunophenotyping of stem cells via FACS/MACS | Isolation of putative CSCs from brain tumors, colon cancer, and HCC [115] [84] [114] |
| Anti-CD44 Antibody | Identification of CSC populations in numerous carcinomas | Critical marker for isolating CSCs in breast, pancreatic, gastric, and colorectal cancers [114] |
| Recombinant EGF & FGF | Growth factors for serum-free culture | Essential components of defined media for in vitro tumorsphere formation assays [84] |
| Hoechst 33342 | DNA-binding fluorescent dye | Used in the Side Population (SP) assay to identify stem cells based on dye efflux capability [115] |
| Verapamil | Inhibitor of ABCG2/BCRP1 transporter | Essential control in SP assays to confirm the specificity of Hoechst dye efflux [115] |
| Lectin MIX (UEA-1/GSL-I) | Binds specific glycan patterns on cell surface | Novel method for detecting CSCs based on glycosylation status in lung and colon cancer [84] |
The strategic profiling of cancer stem cells against their normal counterparts is more than an academic exercise; it is a critical roadmap for developing the next generation of oncology therapeutics. The distinct biological features of CSCs—from their dysregulated signaling pathways and metabolic alterations to their specific cell surface markers—represent a rich source of potential therapeutic windows. Focusing drug discovery efforts on agents that selectively target these CSC-specific vulnerabilities, while sparing normal stem cells, holds the greatest promise for overcoming therapy resistance, preventing metastasis, and achieving durable remissions for cancer patients [117] [114]. The experimental data and methodologies outlined in this guide provide a foundation for this targeted, rational approach to oncogenic potential assessment and therapeutic development.
The development of stem cell-based therapies represents a frontier in modern medicine, particularly in oncology. The regulatory landscape governing these advanced therapies is complex, designed to balance rapid innovation with rigorous safety and efficacy standards. In the United States, the Food and Drug Administration (FDA) provides centralized oversight, whereas globally, frameworks like those from the International Society for Stem Cell Research (ISSCR) offer international guidance that accommodates diverse political, legal, and ethical environments [118]. For researchers and drug development professionals, understanding these frameworks is essential for navigating the pathway from basic research to clinical application, especially when assessing critical risks such as oncogenic potential.
The core challenge in regulation lies in ensuring that stem cell research maintains the highest principles of scientific rigor, oversight, and transparency while translating discoveries into evidence-based therapies [118]. This is particularly crucial when the biological properties that make stem cells therapeutically promising—such as self-renewal, potency, and immunomodulatory capacity—also correlate with potential risks, including malignant transformation [12] [22]. This guide objectively compares the regulatory approaches of the FDA and international frameworks, providing a structured analysis for the scientific community.
FDA Regulatory Framework and Guidelines
Core Regulatory Principles and Oversight
The FDA's Center for Biologics Evaluation and Research (CBER) regulates stem cell-based products under the framework of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps). The agency employs a risk-based approach, focusing on product safety, purity, and potency. For clinical trials, the FDA requires an Investigational New Drug (IND) application, which must comprehensively detail the product's characterization, manufacturing process, preclinical safety data, and proposed clinical protocol. This is especially critical for oncology applications, where the oncogenic potential of cellular products must be thoroughly evaluated before human administration [22].
The FDA emphasizes that the primary societal mission of clinical translation is to alleviate human suffering caused by illness, and this collective effort depends on public support and confidence [118]. The agency's guidance documents, such as the recent draft "Approaches to Assessment of Overall Survival in Oncology Clinical Trials," provide non-binding recommendations that reflect current regulatory thinking. Although this particular guidance focuses on endpoint assessment in randomized trials, its underlying principles of pre-specified safety analysis and robust trial design are fundamental to the evaluation of all novel cancer therapies, including stem cell-based products [119].
Key Considerations for Oncogenic Risk Assessment
The FDA’s regulatory framework requires stringent evaluation of oncogenic risk throughout the product development lifecycle. Key considerations include:
- In Vitro Malignant Transformation: The FDA expects sponsors to assess the risk of spontaneous malignant transformation during in vitro expansion. Studies have shown that extensive culture of Mesenchymal Stem Cells (MSCs) can, in some cases, lead to chromosomal abnormalities and acquisition of tumorigenic properties [22]. The International Society of Cellular Therapy (ISCT) minimal criteria for defining MSCs (plastic-adherence, specific surface marker expression, and tri-lineage differentiation) provide a baseline for product characterization, but standardized long-term safety monitoring protocols are essential [22].
- Tumor Tropism and Microenvironment Interactions: A significant body of evidence demonstrates that exogenously administered MSCs possess a tropism for tumor sites, migrating to tumors via cytokine/receptor pairs such as SDF-1/CXCR4 and PDGF/PDGFr [22]. While this property is exploited for targeted drug delivery, it also necessitates careful evaluation of whether MSCs might inadvertently support tumor progression by modulating the local microenvironment, promoting angiogenesis, or suppressing anti-tumor immune responses [22].
- Long-Term Follow-Up and Monitoring: The FDA often requires long-term follow-up in clinical trials of stem cell therapies to monitor for delayed adverse events, including tumor formation. This is crucial because the immunosuppressive qualities of MSCs may facilitate evasion of the immune system by a nascent tumor, and the consequences of cellular interactions may not be apparent in short-term studies [22].
International Frameworks and Guidelines
The ISSCR Guidelines for Stem Cell Research
The International Society for Stem Cell Research (ISSCR) has established internationally recognized Guidelines for Stem Cell Research and Clinical Translation, which were updated in 2025. These guidelines serve as a global benchmark, promoting an "ethical, practical, and sustainable approach" to stem cell research and therapy development [118] [120]. The ISSCR guidelines are designed to complement existing national laws and are built upon foundational ethical principles, including the integrity of the research enterprise, primacy of patient welfare, and transparency [118].
A key feature of the 2025 update is its targeted revision concerning stem cell-based embryo models (SCBEMs). The update refines oversight recommendations for this rapidly advancing field, retiring previous classifications and introducing the inclusive term "SCBEMs" [120]. It mandates that all 3D SCBEM research have a clear scientific rationale and a defined endpoint, and it explicitly prohibits the transfer of any SCBEM into a human or animal uterus or culturing them to the point of potential viability (ectogenesis) [118] [120]. These provisions are critical for maintaining public trust and defining ethical boundaries in areas of research with complex implications.
Regional Regulatory Landscape: A Case Study of Mexico
A comparison of national regulatory frameworks reveals significant global variation. Mexico's regulatory landscape, overseen by the Federal Commission for Protection against Sanitary Risk (COFEPRIS), offers a contrasting model to the FDA. While the U.S. maintains highly restricted access to non-approved stem cell therapies, Mexico's regulations allow for broader application of certain therapies, particularly those using allogeneic mesenchymal stem cells (MSCs), under a COFEPRIS license [121].
However, this accessibility comes with challenges. Analyses indicate that Mexico's regulatory system has historically contained gray areas and loopholes, which have allowed some private clinics to offer unproven "miracle cures" [122]. The country has been working to strengthen its oversight, including drafting a specific Official Mexican Standard (NOM-260) for stem cells and taking enforcement actions against non-compliant clinics [122]. This landscape makes Mexico a destination for "stem cell tourism," highlighting the practical consequences of divergent international regulatory philosophies on patient access, safety, and the pace of clinical translation [122] [121].
Table 1: Comparison of Key Regulatory Frameworks
| Feature | U.S. FDA Framework | ISSCR International Guidelines | Mexican COFEPRIS Framework |
|---|---|---|---|
| Primary Focus | Safety, efficacy, and quality of products via centralized, binding regulation [119] [121] | Ethical, practical, and sustainable research; international standard-setting [118] [120] | Sanitary authorization and oversight, with evolving specific regulations [122] |
| Key Strength | Rigorous, data-driven review process; high safety standards [121] | Agility in addressing new technologies (e.g., SCBEMs); global consensus-building [120] | Increased patient access to a wider range of therapies [121] |
| Therapy Access | Highly restricted; primarily through clinical trials [121] | Advocates for evidence-based application post-regulatory approval [118] | Broader legal access, leading to medical tourism [122] [121] |
| Oncogenic Risk Mitigation | Required long-term follow-up and extensive product characterization in INDs [22] | Promotes oversight mechanisms, transparency, and rigorous independent peer review [118] | Relies on general health laws; specific norms for advanced therapies are under development [122] |
Comparative Analysis of Key Regulatory Aspects
Approaches to Clinical Trial Design and Endpoint Assessment
The FDA provides detailed recommendations on clinical trial design, emphasizing statistical rigor and the pre-specification of endpoints. In oncology trials, overall survival (OS) is often a key endpoint, and the FDA guides sponsors on its assessment, especially when it is not the primary endpoint [119]. The design must account for the therapy's mechanism of action; for example, a cell therapy with a risk of delayed oncogenesis would necessitate different survival analysis considerations compared to a conventional chemotherapeutic agent.
Internationally, the ISSCR guidelines stress that clinical trials must be "well justified, appropriately designed and ethically sound," with risks that are reasonable in relation to potential benefits [118]. They emphasize the timely exchange of accurate scientific information, including the publication of both positive and negative results, which is vital for the collective understanding of a therapy's long-term risks and benefits [118].
Management of Oncogenic Potential Across the Product Lifecycle
A comparative analysis of how different frameworks address oncogenic risk reveals both parallels and distinctions. The core concerns—malignant transformation during manufacturing, tumorigenicity post-delivery, and long-term consequences—are universal.
Table 2: Oncogenic Risk Assessment Across the Clinical Lifecycle
| Risk Phase | Specific Oncogenic Concern | FDA-Aligned Mitigation Strategies | ISSCR-Aligned Ethical & Oversight Principles |
|---|---|---|---|
| In Vitro Expansion | Spontaneous transformation due to extended culture, chromosomal instability [22] | Strict adherence to defined culture limits, karyotype analysis, and tumorigenicity assays in preclinical models [22] | Research integrity and institutional oversight at each stage [118] |
| Genetic Modification | Insertional mutagenesis, oncogene activation from viral vectors | Extensive testing for vector safety, copy number, and integration sites | Special oversight for irreversible risks, including those from genome editing [118] |
| In Vivo Administration | MSC migration to tumors, support for tumor stroma and metastasis, immune suppression [22] | Biodistribution studies, assessment of impact on established tumors in models | Primacy of patient welfare; protection of vulnerable patients from procedures with no prospect of benefit [118] |
| Long-Term Monitoring | Delayed tumor formation due to persistent cells or altered microenvironments [22] | Requirement for long-term follow-up (e.g., 15 years for gene therapy) in clinical trials | Transparency and sharing of long-term outcome data to build an evidence base [118] |
Experimental Protocols for Oncogenic Assessment
Standardized In Vitro Transformation Assay
Objective: To evaluate the potential for cultured stem cells to undergo spontaneous malignant transformation during extended in vitro expansion. Methodology:
- Cell Culture & Passaging: Expand MSCs (e.g., bone marrow or adipose-derived) under standard conditions, passaging cells upon reaching 80-90% confluence. Maintain a control line cultured to senescence and an experimental line passaged for >20 passages or 4-5 months [22].
- Proliferation Kinetics: Monitor population doubling times and record senescence at each passage. A sudden decrease in doubling time can indicate immortalization.
- Karyotype Analysis: Perform G-banding chromosomal analysis at specific passages (e.g., P5, P10, P15) to detect numerical and structural abnormalities [119].
- Soft Agar Colony Formation Assay: Seed low-passage and high-passage cells in soft agar to assess anchorage-independent growth, a hallmark of transformation. Count colonies after 3-4 weeks [22].
- Telomerase Activity: Measure telomerase activity using the TRAPeze assay. Elevated activity in high-passage cells can suggest a bypass of senescence [119]. Data Interpretation: The emergence of karyotypic abnormalities, sustained telomerase activity, and the ability to form colonies in soft agar are indicative of malignant transformation. The absence of these changes, coupled with normal senescence, suggests a lower risk profile.
In Vivo Tumorigenicity and Biodistribution Study
Objective: To assess the potential of stem cell products to form tumors in vivo and to track their migration and persistence, particularly to tumor sites. Methodology:
- Animal Model: Use immunodeficient mice (e.g., NOD/SCID) to preclude immune rejection of human cells.
- Cell Administration: Administer a high dose of the test stem cells (e.g., 1x10^7 cells per mouse) via a clinically relevant route (e.g., intravenous, intramuscular). Include a positive control (known tumorigenic cells) and a negative control (vehicle).
- Biodistribution Tracking: Label cells with a luciferase reporter or other traceable marker (e.g., GFP) before administration. Use non-invasive bioluminescent imaging at regular intervals (e.g., days 1, 7, 14, 30, 90) to track cell migration and persistence [22].
- Necropsy and Histopathology: At the study endpoint (e.g., 3-6 months), perform a full necropsy. Weigh and preserve major organs (lungs, liver, spleen, bone marrow, brain, gonads). Process tissues for histological analysis (H&E staining) to identify any tumor formation or abnormal growths [22]. Data Interpretation: The formation of tumors at the injection site or in distant organs, particularly those containing human cells confirmed by histology, indicates tumorigenic potential. Biodistribution data revealing significant homing to pre-existing tumors confirms tropism, which must be evaluated for therapeutic benefit versus potential risk.
Visualization of Key Concepts
Oncogenic Risk Pathways of MSCs
The following diagram outlines the primary pathways and mechanisms through which Mesenchymal Stem Cells (MSCs) may contribute to or undergo oncogenesis, from in vitro culture to in vivo effects.
Experimental Workflow for Oncogenic Risk Assessment
This flowchart details a standardized experimental workflow for assessing the oncogenic potential of a stem cell product from the laboratory to preclinical in vivo models.
The Scientist's Toolkit: Key Reagents and Materials
For researchers designing experiments to evaluate the oncogenic potential of stem cells, specific reagents and tools are essential. The following table details key solutions used in the featured experimental protocols.
Table 3: Essential Research Reagents for Oncogenic Potential Assessment
| Reagent/Material | Specific Function | Application in Protocol |
|---|---|---|
| Mesenchymal Stem Cell Media | Supports the growth and maintenance of MSCs in vitro while preserving their undifferentiated state and characteristic marker expression [12]. | In Vitro Transformation Assay; general cell culture expansion. |
| Antibody Panels (CD105, CD73, CD90, CD45, CD34, HLA-DR) | Used in flow cytometry to confirm the immunophenotype of MSCs according to ISCT minimal criteria, ensuring cell population purity and identity before testing [12] [22]. | Cell characterization and quality control at the start of all assays. |
| Soft Agar | Forms a semi-solid medium to test for anchorage-independent growth, a key hallmark of cellular transformation. | In Vitro Transformation Assay (Colony Formation). |
| Luciferase Reporter Construct | Genetically encoded reporter enabling non-invasive, longitudinal tracking of cell location and survival in live animals via bioluminescent imaging. | In Vivo Tumorigenicity and Biodistribution Study. |
| Immunodeficient Mouse Models (e.g., NOD/SCID) | Provide an in vivo environment that does not mount an effective immune response against transplanted human cells, allowing for the assessment of tumorigenicity. | In Vivo Tumorigenicity and Biodistribution Study. |
The translation of stem cell research from laboratory discoveries to clinical therapies represents a revolutionary frontier in modern medicine, offering unprecedented potential to treat a wide range of debilitating diseases and injuries [6]. Within this promising landscape, oncogenic potential assessment stands as a critical safety gateway that must be rigorously evaluated for every stem cell-based product before clinical application. The unique proliferative and regenerative capacities that make stem cells therapeutically valuable also introduce distinct safety challenges, including risks of tumor formation, unintended differentiation, and support of pre-existing cancers [123] [124].
This review examines the comparative oncogenic risks across different stem cell types and analyzes the evolving methodologies designed to mitigate these risks throughout the development pathway. By examining both successful clinical translations and notable setbacks, we aim to provide researchers, scientists, and drug development professionals with evidence-based frameworks for safety assessment. The field must balance the justifiable enthusiasm for regenerative medicine with the scientific rigor required to ensure patient safety, particularly as novel technologies like gene-edited stem cells and complex tissue-engineered products enter clinical testing [124] [6].
Stem Cell Types and Comparative Oncogenic Risk Profiles
Different stem cell types present distinct oncogenic risk profiles based on their origin, potency, and manipulation requirements. Understanding these differences is fundamental to designing appropriate risk mitigation strategies.
Pluripotent Stem Cells: High Proliferative Capacity and Associated Risks
Embryonic Stem Cells (ESCs) and Induced Pluripotent Stem Cells (iPSCs) share the defining characteristic of pluripotency—the ability to differentiate into any cell type in the body. This property makes them incredibly valuable for regenerative applications but also associates them with significant oncogenic risks [123] [6].
The primary concerns with pluripotent cells include:
- Teratoma Formation: The benchmark assay for pluripotency—the ability to form teratomas containing tissues from all three germ layers—also represents a significant safety concern if any undifferentiated cells remain in the final product [123].
- Genomic Instability: Extended in vitro culture of both ESCs and iPSCs can lead to accumulated genetic and epigenetic abnormalities that may predispose cells to malignant transformation [124].
- Insertional Mutagenesis: For iPSCs, the original reprogramming methods using integrating viral vectors raised concerns about oncogene activation, though non-integrative methods have since been developed [6].
Adult Stem Cells: Lower But Not Absent Risks
Mesenchymal Stem Cells (MSCs) and other adult stem cells are multipotent rather than pluripotent, meaning their differentiation potential is limited to specific lineages [123]. While this reduced plasticity generally correlates with lower tumorigenic risk, several concerns remain:
- Culture-Induced Transformation: MSCs maintained in culture for extended periods may undergo spontaneous transformation, with studies reporting the acquisition of genetic abnormalities that could potentially support tumor growth [123].
- Tumor Microenvironment Modulation: Perhaps the most significant concern regarding MSCs is their documented role as carcinoma-associated fibroblasts. When transplanted into a host with undiagnosed cancer, MSCs can home to tumor sites and facilitate cancer progression through immune modulation and trophic support [123].
- Heterogeneity Issues: The inherent heterogeneity of MSC populations, whether endogenous or culture-induced, complicates standardization and safety prediction [123].
Table 1: Comparative Oncogenic Risk Profiles of Major Stem Cell Types
| Stem Cell Type | Source Material | Oncogenic Concerns | Documented Clinical Manifestations |
|---|---|---|---|
| Embryonic Stem Cells (ESCs) | Blastocyst inner cell mass [6] | Teratoma formation, genomic instability during culture, unintended differentiation [123] | No widespread clinical application due to safety and ethical concerns; teratomas documented in preclinical models [123] |
| Induced Pluripotent Stem Cells (iPSCs) | Reprogrammed somatic cells [6] | Teratoma formation from residual undifferentiated cells, genomic instability, insertional mutagenesis from early reprogramming methods [6] | First clinical trials for Parkinson's disease and age-related macular degeneration underway with rigorous tumorigenicity screening [6] |
| Mesenchymal Stem Cells (MSCs) | Bone marrow, adipose tissue, umbilical cord [123] [125] | Culture-induced transformation, facilitation of tumor growth as carcinoma-associated fibroblasts, immune modulation enabling cancer escape [123] | Documented cases of vision loss following adipose-derived MSC injection for macular degeneration; reports of MSC support of breast cancer progression in preclinical models [123] [124] |
| Hematopoietic Stem Cells (HSCs) | Bone marrow, peripheral blood, umbilical cord blood [6] | Limited direct tumorigenicity; potential for occult malignancy transfer in autologous transplantation [6] | Well-established safety profile in thousands of transplants; secondary malignancies primarily associated with conditioning regimens rather than cells themselves [6] |
Methodologies for Oncogenic Risk Assessment
A robust toolkit of experimental approaches has been developed to assess the oncogenic potential of stem cell-based products throughout development.
Core Safety Assessment Protocols
In Vitro Transformation Assays Standardized assays evaluate acquisition of transformation-associated phenotypes including:
- Anchorage-Independent Growth: Soft agar colony formation assays measuring loss of contact inhibition
- Proliferative Control Assessment: Evaluation of serum-independent growth and contact inhibition bypass
- Telomerase Activity Monitoring: Measurement of telomere maintenance mechanisms indicative of immortalization
Genomic Stability Evaluation Comprehensive characterization of genetic integrity includes:
- Karyotypic Analysis: G-banding for chromosomal number and structural abnormalities
- Comparative Genomic Hybridization (CGH): Array-based detection of copy number variations
- Whole Genome Sequencing: Comprehensive identification of point mutations, small insertions/deletions, and structural variants
In Vivo Tumorigenicity Testing The gold standard for safety assessment employs:
- Immunodeficient Mouse Models (e.g., NOD/SCID, NSG): Subcutaneous, intramuscular, or orthotopic implantation with monitoring for 16+ weeks
- Histopathological Analysis: Comprehensive necropsy and tissue examination for teratomas and other neoplasms
- Biodistribution Studies: Tracking cell fate post-transplantation using imaging modalities or PCR-based detection
Diagram 1: Comprehensive Oncogenic Risk Assessment Workflow. This tiered approach progresses from in vitro screening to in vivo testing and long-term clinical monitoring, with decision points at each stage [123] [124] [6].
Advanced Technologies Enhancing Risk Prediction
Recent technological advances have significantly improved the resolution of safety assessment:
Single-Cell RNA Sequencing (scRNA-Seq) This revolutionary technology enables:
- Detection of rare undifferentiated cell populations in differentiated products
- Identification of aberrant transcriptional programs predictive of transformation
- Characterization of heterogeneity in stem cell products that might harbor subpopulations with elevated risk [6]
CRISPR-Based Screening Genome editing technologies facilitate:
- Functional identification of tumor suppressor genes essential for maintaining genomic stability
- Modeling of specific genetic alterations to assess their transformative potential
- Development of reporter systems for monitoring differentiation status and pluripotency [6]
Advanced Imaging Modalities Non-invasive tracking methods include:
- Magnetic particle imaging for long-term cell fate tracking
- Bioluminescence and fluorescence reporters for engraftment and proliferation monitoring
- Multiplexed imaging for histological analysis of potential tumors [6]
Regulatory Frameworks and Quality Control Standards
Regulatory agencies worldwide recognize the unique challenges posed by stem cell-based products and have developed specific frameworks to ensure their safety.
Classification and Oversight Requirements
Stem cell products are typically categorized based on manipulation and intended use:
- Substantially Manipulated Cells: Products that undergo processing that alters their original biological characteristics (e.g., culture expansion, genetic modification) are regulated as drugs/biologics and require full preclinical and clinical testing [124].
- Non-Homologous Use: Cells used for a different basic function than they originally performed require rigorous safety evaluation regardless of manipulation level [124].
Table 2: Essential Research Reagents and Materials for Oncogenic Risk Assessment
| Reagent/Material | Function in Safety Assessment | Key Quality Requirements |
|---|---|---|
| Immunodeficient Mice (e.g., NSG, NOD/SCID) | In vivo tumorigenicity testing; provide permissive environment for human cell growth and potential tumor formation [123] | Defined immune deficiency status, specific pathogen-free conditions, consistent genetic background |
| Cell Culture Media (serum-free, defined components) | Expansion and maintenance of stem cells while minimizing spontaneous differentiation or genetic drift [124] | GMP-grade where possible, documented absence of adventitious agents, lot-to-lot consistency |
| Karyotyping/G-banding Reagents | Detection of chromosomal abnormalities acquired during culture [124] | Standardized protocols, resolution validation, reference standards for abnormal karyotypes |
| Soft Agar Colony Formation Assay Components | Assessment of anchorage-independent growth as a transformation indicator [123] | Optimized agar concentration, positive control cells (known transformed lines), standardized scoring criteria |
| scRNA-Seq Library Preparation Kits | Characterization of cellular heterogeneity and rare undifferentiated populations [6] | High sensitivity for rare cell detection, minimal technical noise, compatibility with downstream bioinformatic analysis |
| Genome Editing Tools (CRISPR/Cas9 systems) | Functional validation of potential oncogenic mutations [6] | High specificity, well-characterized off-target profiles, efficient delivery systems |
Manufacturing and Quality Control
The manufacturing process itself represents a critical control point for oncogenic risk mitigation:
Donor Screening and Cell Banking
- Comprehensive donor medical history and infectious disease testing, particularly for allogeneic products that may be administered to multiple recipients [124]
- Establishment of well-characterized master and working cell banks with defined passage limits to minimize culture-induced genomic changes [124]
Process Controls
- Implementation of Good Manufacturing Practice (GMP) conditions for clinical-grade cell production [124]
- Defined in-process controls and release criteria including viability, identity, purity, and potency assays [124]
- Regular monitoring for genomic stability throughout the manufacturing process [124]
Case Studies in Risk Mitigation: Successes and Setbacks
Successful Clinical Translation with Robust Safety Profiles
Hematopoietic Stem Cell Transplantation As the most established stem cell therapy, hematopoietic stem cell transplantation (HSCT) demonstrates successful long-term risk mitigation through:
- Refined Patient Selection: Comprehensive comorbidity assessment using tools like the Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI) [126]
- Rigorous Donor Matching: HLA matching protocols that minimize graft-versus-host disease while maintaining graft-versus-leukemia effects [6]
- Long-term Monitoring: Established protocols for monitoring secondary malignancies, with studies showing 79% survival rates three years post-treatment [126]
Advances in iPSC-Based Therapies Recent clinical applications of iPSCs highlight evolving safety approaches:
- Non-Integrating Reprogramming: Use of episomal vectors, mRNA, or Sendai virus systems to eliminate insertional mutagenesis concerns [6]
- Comprehensive Genomic Screening: Whole genome sequencing of clinical-grade iPSC lines to exclude lines with oncogenic mutations [6]
- Differential Protox: Development of highly efficient differentiation protocols that minimize residual undifferentiated cells [6]
Notable Setbacks and Lessons Learned
MSC-Associated Adverse Events Case studies illustrate specific risks with MSC therapies:
- Vision Loss Following Intraocular MSC Injection: Reports of patients experiencing severe vision loss after administration of adipose-derived MSCs for macular degeneration highlight the risks of non-homologous use and inadequate safety testing [124]
- Tumor Microenvironment Modulation: Preclinical studies demonstrating MSC integration into tumor stroma and facilitation of cancer progression underscore the importance of excluding patients with active or subclinical malignancies from early-stage trials [123]
ESC-Derived Teratoma Formation Preclinical studies consistently show:
- Dose-Dependent Teratoma Risk: Correlation between number of undifferentiated cells administered and teratoma incidence [123]
- Purity Threshold Requirements: Development of purification strategies to remove undifferentiated cells from differentiated products [123]
Table 3: Quantitative Safety Data from Clinical Stem Cell Applications
| Therapy Application | Patient Population | Oncogenic Safety Events | Risk Mitigation Success Rate |
|---|---|---|---|
| Hematopoietic Stem Cell Transplantation [6] [126] | Blood cancers, genetic disorders | Secondary malignancies primarily associated with conditioning regimens rather than stem cells themselves; minimal direct cellular tumorigenicity [6] | 92% survival rate at 3-year follow-up; 79% overall 3-year survival in recent analyses [126] |
| Mesenchymal Stem Cell Trials (various applications) [123] [124] | Autoimmune diseases, graft-versus-host disease, tissue repair | Isolated reports of ectopic tissue formation; concerns about potential support of pre-existing tumors based on preclinical models [123] | Generally favorable safety profile with ~60-80% success rates in regulated trials; vision loss cases in unregulated settings [124] [126] |
| Pluripotent Stem Cell-Derived Therapies (early-stage trials) [6] | Parkinson's disease, macular degeneration, spinal cord injury | No reported teratomas in initial clinical trials to date, reflecting rigorous purification and safety screening [6] | Early evidence of safety with ongoing monitoring; successful engraftment without tumor formation in initial patients [6] |
Future Directions in Oncogenic Risk Assessment
The field of stem cell safety assessment continues to evolve with several promising developments:
Advanced Prediction Technologies
Humanized Mouse Models Next-generation models with humanized immune systems may provide more predictive assessment of tumorigenicity in immunocompetent environments.
Organoid Co-culture Systems Tumorigenicity assays using complex organoid systems may better recapitulate the human tissue microenvironment for safety assessment.
Liquid Biopsy Monitoring Non-invasive circulating tumor DNA and cell-free DNA analysis for early detection of potential malignant transformation in clinical trials.
Integrated Risk Assessment Frameworks
The future of oncogenic risk assessment lies in multi-parametric approaches that integrate:
- Genomic Data: Comprehensive sequencing with improved bioinformatic prediction of variant pathogenicity
- Functional Data: High-throughput screening of transformation-associated phenotypes
- Clinical Data: Aggregated safety information across trials to identify product-specific risk factors
Diagram 2: Evolution from Current Safety Assessment to Integrated Risk Prediction Frameworks. Future approaches incorporate multi-parametric data and advanced modeling for more predictive safety assessment [123] [124] [6].
The successful clinical translation of stem cell therapies depends fundamentally on robust, comprehensive assessment and mitigation of oncogenic potential. As the field advances toward increasingly complex products—including gene-edited stem cells, tissue-engineered constructs, and combination products—the safety assessment frameworks must similarly evolve. The case studies examined in this review demonstrate that while significant progress has been made in understanding and managing oncogenic risks, continued vigilance and methodological innovation are essential.
The establishment of standardized oncogenic risk assessment protocols, rigorous manufacturing standards, and long-term monitoring frameworks provides a foundation for the responsible translation of stem cell research. By learning from both successes and setbacks, the field can advance toward a future where the remarkable therapeutic potential of stem cells can be realized without compromising patient safety.
Conclusion
The rigorous assessment of oncogenic potential is paramount for advancing stem cell research and therapeutic applications. This synthesis reveals that while foundational understanding of tumorigenic mechanisms has significantly progressed, methodological innovation remains crucial for enhancing detection sensitivity and predictive accuracy. The successful clinical translation of stem cell-based therapies will depend on integrating multifaceted assessment strategies that address cellular heterogeneity, standardize validation protocols, and leverage emerging technologies like single-cell analysis and AI. Future directions should focus on developing personalized risk profiling, establishing universal biomarkers for residual undifferentiated cells, and creating integrated platforms that combine metabolic, genomic, and immunologic profiling to comprehensively evaluate oncogenic risk, ultimately safeguarding the promise of regenerative medicine.
References
- 1. Tumorigenicity as a Clinical Hurdle for Pluripotent Stem Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3967018/]
- 2. Exploring the promising potential of induced pluripotent ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01873-0]
- 3. Cancer stem cells: advances in knowledge and ... - Nature [https://www.nature.com/articles/s41392-024-01851-y]
- 4. Defining the role for adult stem cells as cancer cells of origin [https://pmc.ncbi.nlm.nih.gov/articles/PMC4275355/]
- 5. Induced Pluripotency and Oncogenic Transformation Are ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3528096/]
- 6. recent developments and future prospects in stem-cell therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC11634165/]
- 7. Safety Assessment of Stem Cell-Based Therapies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12609812/]
- 8. Cancer stem cells: landscape, challenges and emerging ... [https://www.nature.com/articles/s41392-025-02360-2]
- 9. Targeting self-renewal pathways in cancer stem cells [https://www.nature.com/articles/oncsis201535]
- 10. Self-renewal and differentiation in squamous cell carcinomas [https://pmc.ncbi.nlm.nih.gov/articles/PMC6834421/]
- 11. Embryonic Stem Cells: Controversy, Mechanisms, and ... [https://www.dvcstem.com/post/embryonic-stem-cells]
- 12. Mesenchymal stem cells in treating human diseases [https://www.nature.com/articles/s41392-025-02313-9]
- 13. Balancing hematopoietic stem cell self-renewal and ... [https://www.sciencedirect.com/science/article/pii/S0301472X25001110]
- 14. Proteomic-based stemness score measures oncogenic ... [https://www.sciencedirect.com/science/article/pii/S2666979X25001077]
- 15. Oncogenesis and cancer stem cells - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4515053/]
- 16. Cancer Stem Cells—Origins and Biomarkers: Perspectives ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.01280/full]
- 17. The dual role of autophagy in cancer stem cells [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06595-z]
- 18. Tumor microenvironment of cancer stem cells [https://www.sciencedirect.com/science/article/pii/S2352304223003240]
- 19. Biomarkers of Cancer Stem Cells for Experimental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9147761/]
- 20. UCLA scientists reprogram stem cells to create renewable ... [https://www.uclahealth.org/news/release/ucla-scientists-reprogram-stem-cells-create-renewable-cancer]
- 21. ESMO 2025: MSK Presents New Research, Including ... [https://www.mskcc.org/news/esmo-2025-msk-presents-new-research-including-latest-on-lung-and-pancreatic-cancer-treatments]
- 22. The Oncogenic Potential of Mesenchymal Stem Cells in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2873198/]
- 23. Stem cell mutations, associated cancer risk, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10624213/]
- 24. The mutational impact of culturing human pluripotent and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7237696/]
- 25. Tissue-specific mutation accumulation in human adult stem ... [https://www.nature.com/articles/nature19768]
- 26. Low rates of mutation in clinical grade human pluripotent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7089967/]
- 27. Somatic mutation and selection at population scale [https://www.nature.com/articles/s41586-025-09584-w]
- 28. Human induced pluripotent stem cells display a similar ... [https://www.sciencedirect.com/science/article/pii/S2589004222000062]
- 29. Stem Cells and the Niche: A Dynamic Duo - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3012646/]
- 30. Perspective The Stem Cell Niche in Regenerative Medicine [https://www.sciencedirect.com/science/article/pii/S193459091200077X]
- 31. Cancer stem cells and niches: challenges in ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02265-2]
- 32. Cancer stem cells: Bridging microenvironmental interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12264091/]
- 33. Stem Cell for Cancer Immunotherapy: Current Approaches ... [https://link.springer.com/article/10.1007/s12015-025-10933-5]
- 34. Patient-derived xenograft models: Current status, challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12179623/]
- 35. and immunocompetent Immunocompromised for... mouse models [https://www.dovepress.com/immunocompromised-and-immunocompetent-mouse-models-for-head-and-neck-s-peer-reviewed-fulltext-article-OTT]
- 36. Immunocompetent murine models for the study of glioblastoma... [https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-12-107]
- 37. Innovative organ-on-a-chip platforms for exploring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12269115/]
- 38. Advancing In Vivo Detection of T-Cell Function: Development ... [https://jnm.snmjournals.org/content/early/2025/07/24/jnumed.125.269799]
- 39. Three-Dimensional Cell Cultures: The Bridge between In ... [https://www.mdpi.com/1422-0067/24/15/12046]
- 40. Comparative analysis of 3D-culture techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12307937/]
- 41. Novel 3D Liquid Cell Culture Method for Anchorage ... [https://www.nature.com/articles/s41598-018-21950-5]
- 42. How Are Organoids Different from 3D Primary Cell Cultures? [https://blog.crownbio.com/how-are-organoids-different-from-3d-primary-cell-cultures]
- 43. Examining the Market for Organoid Culture Systems in 2025 [https://eureka.patsnap.com/report-examining-the-market-for-organoid-culture-systems-in-2025]
- 44. The use of stem cells and organoids for modeling host- ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1641366/full]
- 45. Exploring the Potential of Stem Cell Treatments (2025) [https://www.dvcstem.com/post/stem-cell-treatment]
- 46. Markers and Reporters to Reveal the Hierarchy in ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.668851/full]
- 47. toward enrichment of cancer stem cells - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12008981/]
- 48. Reporter gene systems for the identification and ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8316869/]
- 49. Flow Cytometry and Cell Marker Analysis in Cancer ... [https://www.marinbio.com/flow-cytometry-and-cell-marker-analysis-in-cancer-diagnosis-prognosis-and-treatment/]
- 50. Flow Cytometry Analysis of Mesenchymal Stem Cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC12118590/]
- 51. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 52. Stemness signatures reflect prognostic disturbances in gastric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278261/]
- 53. CD51 promotes gastric cancer stemness via blocking ... [https://www.sciencedirect.com/science/article/pii/S0304383525004549]
- 54. Diagnostic value of expressions of cancer stem cell ... [https://www.spandidos-publications.com/10.3892/ol.2024.14669]
- 55. Induced Pluripotent Stem Cell (iPSC) Industry Trends for ... [https://bioinformant.com/ipsc-industry-trends/]
- 56. Clinical translation of human iPSC technologies: advances ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1627149/full]
- 57. Next-Gen Therapeutics: Pioneering Drug Discovery with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12011342/]
- 58. Recent Advances in High-Content Imaging and Analysis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10572296/]
- 59. Drug discovery research with the iPSC models of ... [https://www.sciencedirect.com/science/article/pii/S0168010225001683]
- 60. Cell-based drug discovery | Why human iPSC-derived ... [https://www.bit.bio/blog/cell-based-drug-discovery-with-human-ipsc-models]
- 61. How Human iPSCs are Revolutionizing Drug Discovery in ... [https://bioinformant.com/ipsc-drug-discovery/]
- 62. iPSC-based Platforms Market 2025 Report and Trends [https://www.towardshealthcare.com/insights/ipsc-based-platforms-market-sizing]
- 63. Advancing high-throughput cardiac screening with ... [https://www.ddw-online.com/advancing-high-throughput-cardiac-screening-with-physiologically-relevant-3d-ipsc-based-models-34465-202504/]
- 64. Transforming microfluidics for single-cell analysis with robotics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12587405/]
- 65. Artificial intelligence and systems biology analysis in stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12476622/]
- 66. Systematic comparison of high-throughput single-cell RNA- ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014425000627]
- 67. Concepts and new developments in droplet-based single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11568944/]
- 68. Single Cell Genomics Market Research 2025 [https://aijourn.com/single-cell-genomics-market-research-2025-revolutionizing-genomics-out-of-the-lab-researchandmarkets-com/]
- 69. Microfluidics and Single-Cell Analysis - Medical Micro Molding [https://www.medicalmicromolding.com/microfluidics-and-single-cell-analysis-revolutionizing-precision-biology/]
- 70. Microfluidics applications for high-throughput single cell ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-021-01045-6]
- 71. Application of artificial intelligence-based stemness index ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380755/]
- 72. A machine learning and explainable artificial intelligence ... [https://www.sciencedirect.com/science/article/pii/S2772442523000370]
- 73. Single-cell Omics Industry Automation 2025: How AI ... [https://www.linkedin.com/pulse/single-cell-omics-industry-automation-2025-how-ydv9e/]
- 74. Highly Sensitive Detection of Human Pluripotent Stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9622575/]
- 75. High throughput screening of mesenchymal stem cell lines ... [https://www.nature.com/articles/s41598-022-21653-y]
- 76. Purity Assessment by Flow Cytometry for Lineage-Specific ... [https://www.stemcell.com/purity-assessment-by-flow-cytometry-lineage-specific-chimerism-analysis-lp.html]
- 77. 髓系肿瘤体细胞变异检测中参考样本类型的优化评估 [https://www.ebiotrade.com/newsf/2025-10/20251022212640288.htm]
- 78. Single-cell multi-omics and machine learning for dissecting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570030/]
- 79. Review Article Cancer cell cycle heterogeneity as a critical ... [https://www.sciencedirect.com/science/article/pii/S2352304223000041]
- 80. Stem Cells in Cancer: From Mechanisms to Therapeutic ... [https://www.mdpi.com/2073-4409/14/7/538]
- 81. A Guide to Reproducibility in Preclinical Research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6314499/]
- 82. Comparability and reproducibility of biomedical data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3713713/]
- 83. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4756645/]
- 84. Comparison of methods for cancer stem cell detection in ... [https://www.nature.com/articles/s41416-024-02839-9]
- 85. and Standardized measurement of decision-making in... reproducible [https://elifesciences.org/articles/63711]
- 86. breast cancer patients: a retrospective study - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12290898/]
- 87. Exploring Genomic Assays for Early-Stage Breast Cancer ... [https://www.targetedonc.com/view/exploring-genomic-assays-for-early-stage-breast-cancer-treatment]
- 88. First versus second-generation molecular profiling tests [https://www.sciencedirect.com/science/article/pii/S0305737225000313]
- 89. Elimination of tumorigenic pluripotent stem cells from their ... [https://www.sciencedirect.com/science/article/pii/S221367112500147X]
- 90. Pluripotent Stem Cells: Recent Advances and Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12025069/]
- 91. Tracing genomic instability in induced mesenchymal ... [https://www.nature.com/articles/s12276-025-01439-8]
- 92. Evaluating teratoma formation risk of pluripotent stem cell- ... [https://www.sciencedirect.com/science/article/pii/S146532492500684X]
- 93. Precision Reprogramming: How AI Tricks Cancer's Toughest ... [https://today.ucsd.edu/story/precision-reprogramming-how-ai-tricks-cancers-toughest-cells]
- 94. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 95. The role of stem cells in precision medicine: next-generation ... [https://jenci.springeropen.com/articles/10.1186/s43046-025-00328-5]
- 96. A risk-based approach for cell line development ... [https://www.sciencedirect.com/science/article/pii/S1465324922007460]
- 97. Improving tumor microenvironment assessment in chip ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1462293/full]
- 98. A Preclinical Pipeline for Translational Precision Medicine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8472761/]
- 99. Stem cells as biological drugs for incurable diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12505543/]
- 100. Drivers and implications of lineage-specific cancer-related ... [https://www.sciencedirect.com/science/article/pii/S2213671125002474]
- 101. Human Pluripotent Stem Cell-Derived Alveolar Organoids for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594594/]
- 102. In vitro-In vivo Correlation: Perspectives on Model Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC3099258/]
- 103. Explaining in-vitro to in-vivo efficacy correlations ... [https://pubmed.ncbi.nlm.nih.gov/37930506/]
- 104. CMC Perspectives of In Vitro / In Vivo Correlation (IVIVC) ... [https://premier-research.com/perspectives/cmc-perspectives-of-in-vitro-in-vivo-correlation-ivivc-in-drug-development/]
- 105. Drug delivery to solid tumors: the predictive value of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5673046/]
- 106. In Vitro 3D Models of Haematological Malignancies [https://www.mdpi.com/2073-4409/14/1/38]
- 107. Human Bone Marrow Organoids for Disease Modeling ... [https://pubmed.ncbi.nlm.nih.gov/36351055/]
- 108. Explaining in-vitro to in-vivo efficacy correlations in ... [https://link.springer.com/article/10.1007/s10928-023-09891-7]
- 109. Explaining in-vitro to in-vivo efficacy correlations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10982099/]
- 110. Normal stem cells and cancer stem cells: similar and different [https://pubmed.ncbi.nlm.nih.gov/20435143/]
- 111. Crucial differences between cancer and normal stem cells [https://ecancer.org/en/news/3063-crucial-differences-between-cancer-and-normal-stem-cells]
- 112. Convergence of normal stem cell and cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3192222/]
- 113. Therapeutic Window, a Critical Developmental Stage for Stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6546416/]
- 114. Targeting cancer stem cells for reversing therapy resistance [https://www.nature.com/articles/s41392-020-00430-1]
- 115. Characterization of normal and cancer stem cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC5410217/]
- 116. Biomarkers and targeted therapy for cancer stem cells [https://www.sciencedirect.com/science/article/abs/pii/S0165614723002584]
- 117. Targeting of human cancer stem cells predicts efficacy and ... [https://www.sciencedirect.com/science/article/pii/S0304383524005032]
- 118. Guidelines — International Society for Stem Cell Research [https://www.isscr.org/guidelines]
- 119. Overall Survival in Oncology Clinical Trials [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/approaches-assessment-overall-survival-oncology-clinical-trials]
- 120. The ISSCR Releases Targeted Update to the Guidelines ... [https://www.isscr.org/isscr-news/the-isscr-releases-targeted-update-to-the-guidelines-for-stem-cell-research-and-clinical-translation]
- 121. Stem Cell Therapy USA vs. Mexico (2025) - Cost & Safety ... [https://www.placidway.com/article/5430/Stem-Cell-Therapy-in-the-USA-vs-Mexico-Cost-Quality-and-Accessibility]
- 122. SCA LRA Update - April 2025: Regulatory Landscape of ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/04/09/sca-lra-update-april-2025-regulatory-landscape-of]
- 123. A Review of Stem Cell Translation and Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3872439/]
- 124. 3. Clinical Translation of Stem Cell-based Interventions [https://www.isscr.org/guidelines/clinical-translation-of-stem-cell-based-interventions]
- 125. Stem Cell Research: The Future of Regenerative Medicine ... [https://www.dvcstem.com/post/stem-cell-research]
- 126. Stem Cell Therapy Success Rates Hit 78%: New Research ... [https://globalrph.com/2025/03/stem-cell-therapy-success-rates-hit-78-new-research-reveals-breakthrough-results/]